

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Precision medicine for common chronic diseases entails the customization of patient care based on genetic, clinical, and environmental information sourced from large populations [1]. Risk prediction models for chronic diseases are useful tools for classifying high-risk groups that require educational or clinical therapeutic interventions. However, traditional risk factors for common chronic diseases do not typically appear early in life, making it difficult to fully identify individuals at high-risk. Genome-level analyses are attractive in that they provide predictive information about the entire trajectory of a disease at an early stage of life; previous studies have reported that around 40% to 50% of phenotypic variance in susceptibility to chronic diseases such as coronary artery disease (CAD) or type 2 diabetes (T2D) can be explained by genetic factors [2,3]. However, over the last decade, genome-wide association studies (GWAS) have demonstrated that each individual genetic variant generally has only a small or modest effect. Thus, from the current perspective, the most common complex diseases that pose public health concerns are highly polygenic in nature, with hundreds or thousands of small-effect genetic variants influencing the development or progression of the disease [4].
This situation has led to the development of models for polygenic risk scoring, which aggregate the effects of multiple single-nucleotide polymorphisms (SNPs) into a single score. More specifically, polygenic risk scores (PRSs) represent a method of aggregating an individual's genetic information, weighted by the associations of genetic variants with disease outcomes as identified from GWAS, into simplified scores that capture an individual’s susceptibility to disease and can therefore be used for risk prediction [5]. Previously, PRS models were based on a small number of SNPs identified through candidate gene studies and scores calculated simply by summing an individual’s risk allele burden for a handful of disease-associated variants, all weighted equally regardless of the underlying strength of association. Recent advances in the organization of large-scale consortia and biobanks have enabled the brisk discovery of genetic variants associated with common complex diseases [6–8]. In addition to these large datasets becoming available, computational and statistical advances have allowed easier derivation, calculation, and validation of PRS models [9]. This has led PRSs to be widely applied in research studies, consequently confirming the genetic contributions of common variants to disease status [3,10]. This review aims to briefly introduce the methods of calculating a PRS, examples of clinical utility for cardiometabolic PRSs, and considerations for the application of PRSs in clinical practice.
Go to : 
THE PROCESS OF CREATING POLYGENIC RISK SCORES
Before discussing the clinical utility of PRS profiling, we briefly introduce the process by which PRSs are developed and evaluated. PRSs are calculated through statistical models, which are constructed via the five-step process outlined below [11].
1) Selection of base and target datasets
2) Single-nucleotide polymorphism selection and weight calculation: First, a base GWAS dataset is needed that provides summary statistics, including beta coefficients and the P-values of genotype and phenotype associations. This dataset is used to identify disease-associated SNPs and their effect sizes. Weighting parameters that affect the calibration and predictive ability of the final model are also determined from the base dataset. In addition, a target dataset consisting of genotypes and phenotypes is required, which should be independent of the base dataset as it is used for calculating individual PRSs and checking the overall performance accuracy.
3) Dataset quality control: To ensure high accuracy and validity when performing a PRS analysis, quality control of both the base and target datasets is very important. Regarding genotype and phenotype data, the quality control process includes addressing missing SNPs and individuals, checking for sex discrepancies, including only SNPs above an appropriate minor allele frequency threshold, and excluding individuals on the basis of relatedness and high or low heterozygosity rates.
4) Shrinkage of GWAS effect size estimates and controlling linkage disequilibrium: Since not all SNPs influence the trait under study, using the unadjusted effect size estimates of all SNPs could lead to poor prediction outcomes with high standard error. To avoid this problem and control for linkage disequilibrium, two methods for shrinking the effect size estimates are broadly used: (1) statistical shrinkage/regularization techniques such as Lasso or ridge regression, or Bayesian approaches that perform shrinkage via prior distribution specification, and (2) a P-value selection threshold with clumping. In some cases, using a threshold below that for genome-wide statistical significance may improve performance, often at the expense of generalizability. PRSs can be readjusted with consideration of effect size biases, including the effect size inflation typical in a base dataset, the presence of multiple linked variants within each disease-associated locus, subphenotypes of interest, and ethnic or demographic factors that may influence generalizability.
5) Generation and validation of PRSs: PRSs can be generated by a mathematical algorithm that combines selected SNPs, assigns weighting based on effect size, and determines the best-performing SNPs in the population of the independent target dataset. Separate populations should be used to confirm the validity of a PRS, and considerations that should be taken into account include the PRS unit, population genetic structure, PRS distribution, and overfitting.
Go to :
GENETIC EFFECTS OF COMMON VARIANTS ON COMMON COMPLEX DISEASES
The clinical utility of GWAS-based inherited risk estimates has mainly been assessed in terms of discriminative ability as described by the area under the curve, sensitivity, and specificity [5,12,13]. The area under the curve is a population-level metric reflecting the overall probability of discriminating individuals with a given disease from those without [14]. This metric cannot provide information regarding the absolute predictive risk conferred to a single individual or subgroup. In contrast, a PRS can identify a subset of individuals whose absolute risk of disease is significantly higher than that of the average individual in the general population. One of the major utilities of PRSs may be the comprehensive stratification of an overall population through accounting for each individual's or subgroup's respective genetic susceptibility [15].
In previous papers, PRSs have demonstrated considerable performance in predicting target diseases. Khera et al. [5] showed that, in five common diseases, including CAD and T2D, genome-wide PRSs could identify a larger fraction of the population than could carrier status for rare monogenic mutations, while considering a comparable risk level. With respect to CAD in particular, individuals in the top 1% of the PRS distribution had 4.8-fold higher risk than the remaining 99%. In addition, relative to the carrier frequency of familial hypercholesterolemia, which is caused by a rare monogenic mutation, the PRS identified 20-fold more individuals at comparable risk. Those high-risk individuals could benefit from tailored health management strategies, which may include intensive screening or more invasive preventive interventions. Another study suggested that, compared to individuals with average PRSs, those in the top 2.5 percentile experienced the onset of common diseases 4.3 to 6.6 years earlier [13]. They also evaluated the effect of adding the PRS to clinical risk scores on discriminative and reclassification performance and found that the addition of PRS significantly enhanced both the concordance index and net reclassification improvement.
Genetic risk may be modifiable through adherence to a favorable lifestyle or medication. Previous studies have demonstrated the effects of PRSs, lifestyle factors, and their interaction on the risk of chronic metabolic diseases. In particular, Khera et al. [16] suggested that genetic and lifestyle factors are independently associated with CAD risk. Even in individuals at high genetic risk, a favorable lifestyle reduced the relative risk of CAD by nearly half relative to an unfavorable lifestyle. The log additive effects of combined genetic composition and lifestyle behaviors on disease risk have also been reported in atrial fibrillation, stroke, hypertension, and T2D [17].
Go to :
CLINICAL UTILITY OF POLYGENIC RISK SCORES
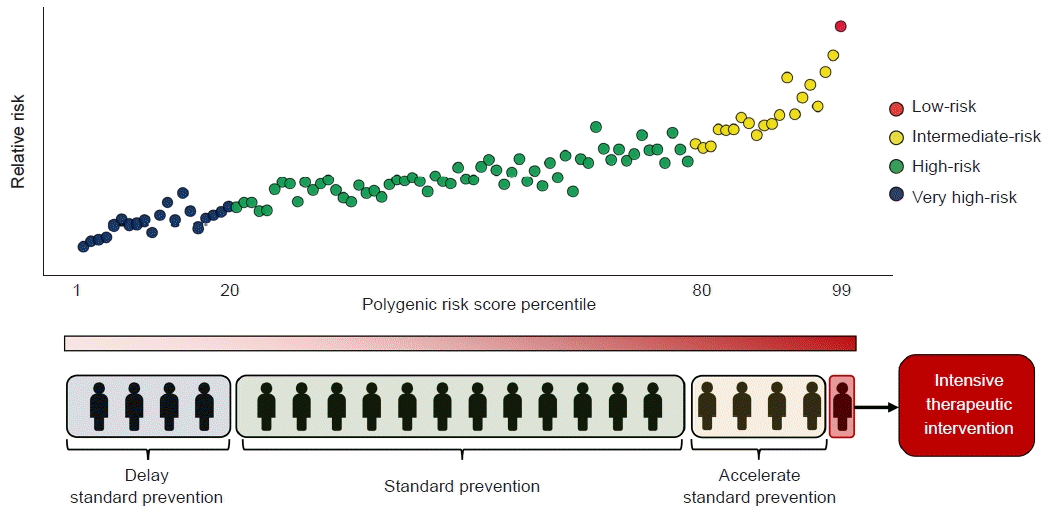
Combining PRSs with traditional clinical risk factors can further refine prevention strategies by enabling more sophisticated risk stratification (Fig. 1). Although PRSs are not yet routinely used in clinical practice, they are proposed to have several advantages. First, PRSs can be ascertained from birth, and only one measurement is needed to determine the score. Cardiovascular risk captured by PRSs appears to be largely independent of traditional risk factors, which usually manifest in middle-aged adults [18]. Thus, lifestyles can be optimized from infancy and genetic information can potentially influence motivation for disease prevention from very early in a patient’s life. Second, PRSs may be appropriate for screening common metabolic diseases in large community populations, especially in young adults. Several results have demonstrated their utility in decisions for early or late initiation and the interpretation of disease screening (e.g., for breast, colorectal, or prostate cancer) [19–21]. Third, PRSs can affect the disease course or progression as well as disease incidence. We previously demonstrated that individuals with high PRSs for CAD and T2D had significantly higher risk of cardiovascular mortality, the most progressive manifestation of CVD [22]. Fourth, PRSs can factor in target selection for interventions to treat or prevent disease. Individualized treatment is central to precision medicine, and applying PRSs to improve individual patient care would benefit the decision-making process for diagnosis and treatment and thereby guide therapeutic interventions. A high PRS may assist in triaging individuals in a borderline risk group. In the context of cardiovascular disease, there has been considerable debate as to whether PRSs are appropriate as interventions for primary preventive treatments, such as antiplatelet drugs and statins. For example, in individuals with borderline low-density lipoprotein cholesterol levels, a CAD PRS in addition to traditional risk factors may permit a more detailed categorization of risk, thus influencing the decision to initiate statin therapy [23,24].
Go to :
FUTURE DIRECTIONS
PRSs offer a valuable opportunity to improve the early identification of actionable cardiovascular risk. Nonetheless, despite encouraging findings, the clinical utility of PRS risk estimation remains limited. Most studies have primarily focused on individuals of European ancestry, and generalization to other ancestries might be difficult due to linkage disequilibrium. In addition, evidence relating to PRS application is still lacking, and many models based on a PRS alone still have much lower area under the curves than traditional multivariate models, as expected from the use of a single risk factor due to several limitations. Thus, further research should be conducted, especially more large-scale prospective studies examining the clinical utility of PRSs, and caution remains needed when interpreting PRS results and applying them to the general population. The dissemination of false deterministic beliefs should be rejected. As methodological advances increase the accuracy of PRS determinations and so continue to improve PRS estimates, it is expected that PRSs will become more useful.
Go to :
 XML Download
XML Download