

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The primary goals of medical research include the identification of progression, specific consequences, and risk factors of a disease.1) In this regard, a randomized controlled trial (RCT) is required to establish a causal relationship between exposure and outcome, and therefore, it is the most representative study design method when conducting medical research. However, an RCT cannot always be performed; consequently, many medical studies are observational instead.
In observational studies, it can be difficult to rule out the effects of confounding variables between exposure and outcomes, and there is a possibility of false causal inferences, regardless of the use of an appropriate study design and statistical methods.2) To reduce these errors, the instrumental variable (IV) method has been proposed as an alternative statistical method for investigating the causal relationship between exposure and outcome, while controlling for confounding variables. The IV method was first introduced by econometricians and later applied in Mendelian randomization (MR) analysis in medical statistics. The MR approach was suggested by Katan in 1986,3) wherein it was explained how various apolipoprotein E isoforms could be used as IVs for investigating the association between serum cholesterol levels and cancer risk.4) However, MR research did not start to gain popularity until 2011; in 2015, a report on MR was published in a special issue of the International Journal of Epidemiology. In the same year, a book regarding MR was also published.5) Nevertheless, many researchers remain uncertain of the approach toward MR studies. Here, we introduce the basic concept of MR, covering analysis and extension methods (Table 1).
BASIC CONCEPT
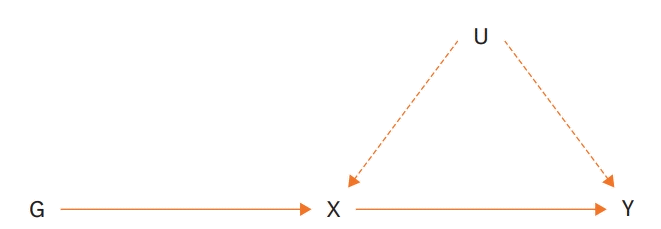
MR was derived from the concept of genetic variants randomly assigned according to Mendel's second law.1) Whereas RCTs are randomized to control for confounders during the clinical trial period, in MR studies, genes are assigned to individuals prior to exposure to other factors. Since these genetic factors cannot be modified, genetic variants, such as single-nucleotide polymorphisms (SNPs), are used as IVs for MR analysis.2)6-8) The general aim of the MR approach is to estimate the causal effect of an exposure (X) on an outcome (Y) using genetic variants (G) for X (Figure 1).9)
Two-stage least squares regression
Two-stage least squares (2SLS) is a two-step method that uses continuous outcomes and a linear model.10)11) The first step estimates the effect on exposure () to the instruments, and the second step estimates the effect on the outcome (yi) through the estimated exposure (). Thus, the variation in exposure described by IV identifies the effect on the outcome, and the causal estimate is reflected as a regression coefficient (βIV) for the change in outcome due to the unit change in exposure.12)
For example, let us suppose that we have an IV available.11) With data on an individual indexed by i=1, ⋯, N who have exposure xi and outcome yi and assuming an additive linear model for the IVs gik indexed by k=1, ⋯, K, the first-stage regression model is represented as follows:
The fitted values are then used in the following second-stage regression model:
where εXi and εYi are independent error terms. If both models are estimated by standard leastsquares regression, both the error terms are implicitly assumed to be homoscedastic and normally distributed. Estimating the causal effect in a 2-stage method provides the correct point estimate; however, uncertainty in the first-stage regression is not considered. Thus, the standard error from the second-stage regression is not correct.13)
If the genetic instrument is a path from X to Y, the direct effect of the instrument on the outcome Y (βGY) is equal to the product of the effects on the pathway mediated by exposure (i.e., βGY=βGX×). Accordingly, the causal effect can be estimated by dividing the effect of IV on the outcome (βGY) by the effect of IV on the exposure (βGX) as follows.9)
Since the formula is calculated as the ratio of 2 IV-based effect estimates, it is also called a ratio estimate or Wald estimate. The variance of is estimated through the delta-method based on Taylor series expansion and can be approximated as follows:
Approximations can be easily identified through statistical programs, such as R, SAS, or STATA.
The 2SLS regression method is also applicable when outcome Y is binary. In this case, an approximate normal distribution of X is required, and the causal relative risk or odds ratio parameter can be estimated using a log-linear or logistic regression model in the second-stage regression.11) However, even in binary outcomes, uncertainty in the first-stage regression is not accounted for, such that the standard error of first-stage coefficients has slightly less precision. This can be solved using a likelihood-based or bootstrap method.14) The 2SLS method using a non-linear second-stage regression model has been criticized for being called “forbidden regression” because it does not guarantee un-correlation of residuals and IVs in the second-stage regression.14)15) Debate on the interpretation and validity of such estimates is ongoing.11) In addition to 2SLS, a limited information maximum likelihood method is available for calculating the IV estimate,16) although here, we have only explained 2SLS.
ASSUMPTIONS OF MR
Multiple IVs can be used when conducting MR studies via 2SLS. In this case, rather than choosing an IV that is likely to be associated with unconditional research, it is necessary to meet the assumptions. To infer correct causality, the choice of genetic IV in MR studies must be carefully considered, and the 3 main assumptions for allowing IV are as follows.1)8)15)17-19)
1) The genetic variant is strongly associated with the exposure.
2) The genetic variant is independent of the outcome, given the exposure and all confounders (measured and unmeasured) of the exposure-outcome association.
3) The genetic variant is independent of factors (measured and unmeasured) that confound the exposure-outcome relationship.
These assumptions cannot be easily tested because not all confounders are observed, although they should be confirmed based on the subject matter or background knowledge.18) Assumption 1 confirms the degree of association between the IV and exposure. F-statistics and R2 are commonly used for identification.12) The relationship between the genetic variants selected for exposure and exposure can be confirmed by linear regression. For F-statistics >10, there is a strong association between genetic variants and exposure.20) Additionally, only genetic variants with p-values <5×10−8 are used for analysis according to genotype using Cuzick's test for trend.16)21)22) Assumption 2 can be demonstrated by showing that IV affects outcome through exposure, although it can be difficult to verify the same directly due to SNPs in linkage disequilibrium or horizontal pleiotropy of SNPs.1) However, the assumption that there is no association between the IV and confounder owing to random allocation of alleles is often difficult to prove indirectly by empirically evaluating the association. Finally, Assumption 3 is also difficult to directly prove due to pleiotropy. Indirect tests, including the Sargan and Hansen tests, analyze over-identifying restrictions,23) identifying the residual effects of genetic instruments on an outcome.
GENETIC RISK SCORES
If the analysis is based on multiple genes and it is known that different biological pathways function between the genes and traits, it is important to include all related information. Using multiple IVs, rather than one IV, can help solve weak instrument bias. Genetic risk score (GRS) is used to enhance the quantitative effect of IVs on risk factors.24) GRS analysis is based on SNPs selected from genetic information analysis, and SNPs included in the analysis are strongly associated with the exposure and are stratified for low linkage disequilibrium.
There are 2 methods of GRS, namely, counted GRS (cGRS) and weighted GRS (wGRS). cGRS is a simple method of adding the number of risky alleles in each SNP, while weighted GRS first multiplies the weight and number of risky alleles of each SNP and then adds them. For a multi-SNP risk score depending on k chosen SNPs, the value of the risk score for the i-th subject is as follows:
where xij is the dose of the coded allele at the k-th SNP in the i-th subject and wk is a chosen coefficient or weight for the k-th SNP.25) The GRS obtained in this way fits the first-stage regression model as an IV in 2SLS.
EXTENSIVE MR
The MR method using 2SLS is a standard for estimating one-sample MR or single-sample MR. However, some extensions to the MR approach have been developed in recent years. In the following sections, we introduce two-sample MR and bidirectional MR; in addition, 2-step MR, multivariable MR, and factorial MR have been developed.1)2)19)
Two-sample MR
Standard MR is analyzed using only one-sample data. However, 2-sample MR is a method for estimating the causal effect when exposure and outcome data are measured in different samples.2)26)27) This can be used when it is difficult or expensive to measure both exposure and outcome for the same data. In addition, 2-sample MR has become increasingly popular as the scope of analysis has been extended by using summary data of publicly available genomewide association studies (GWAS).28)
Bidirectional MR
Bidirectional MR can determine whether exposure causes an outcome or whether an outcome causes exposure.29) The study is first conducted in the direction of exposure to outcome and then in the opposite direction. This method determines whether exposure affects outcomes in the opposite direction or by potential confounding between exposure and outcome.30) Nonetheless, the complexity of biological systems may make interpretation of such analytical results difficult.17)
LIMITATION
The first limitation of MR is that it requires large sample sizes.31) In many cases, genetic variant proxying for exposure or traits can only account for a very small proportion of the variance in exposure or traits. To obtain an accurate risk estimate, thousands of samples are generally needed. The second limitation of MR is population stratification. Spurious associations may arise in MR where the genetic variant and outcome are associated with ancestral background in an admixed or stratified sample.2) To address limitations, there are methods for performing analyses only on homogenous populations or for controlling populations appropriately using principal components analysis or linear mixed models. The third limitation of MR is winner's curse. In the case of single-sample MR, if the discovery GWAS and MR analysis for the genetic instrument use the same sample, the IV and exposure estimates may be biased upward.26) This can be averted by using the aforementioned 2-sample MR. The final limitations are trait heterogeneity, horizontal pleiotropy, and linkage disequilibrium. These limitations break existing MR assumptions and require understanding of genetic variants and biological knowledge. Special methods for detecting pleiotropy include MR-Egger regression32) and weighted median approaches.33)
CONCLUSION
When conducting MR analysis, it is important to be aware of the validity of the assumptions supporting the study and how previous studies have been reported. The most important aspect of MR is the choice of IV.19) When conducting studies with some considerations clarified, these findings potentially provide unbiased information on exposure and IV, which can then be used to assess new causal relationships or verify the results studied in RCTs. MR will be applied further in the future as a statistical method to identify causal effects. In addition, the extension of the MR approach may provide a potentially fruitful method for strengthening causal inference in epigenetic studies, and these tools can be applied to contemporary large-scale epigenetic studies. Therefore, efforts must be made to overcome the limitations of MR analysis to ensure precise studies.
 XML Download
XML Download