

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
CONCEPT OF PCTs
Table 1.
![]()
Participants: ECTs use high levels of inclusion and exclusion criteria to select the appropriate research targets. However, PCTs minimize the exclusion criteria to include a variety of patients visiting the routine clinical sites.
Settings: ECTs are performed in strictly controlled settings to minimize bias. By contrast, PCTs are performed in multiple institutions that cover the full range of actual clinical trials, not in a single center, to maximize applicability and generalizability.
Intervention: ECTs provide specific directions, so that the delivery of intervention and compliance with intervention are closely monitored. However, PCTs leave the providers with details of the implementation of intervention as in the normal practice and do not require compliance with the interventions.
Comparator: ECTs recommend that the therapeutic impact of experimental interventions be compared with placebo, but PCTs generally compare the relative effectiveness of the treatment studied with the treatment currently used in practice instead of the comparing it with a placebo.
Outcomes: PCTs focus on a broad set of patient-centered outcomes or endpoints; however, ECTs have limited or surrogate measurable outcomes, especially clinical symptoms or biological markers. Consequently, PCTs have high relevance to real life and direct social value than ECTs because they are more likely to be applied to practice at once.17) Based on these factors, Califf and Sugarman11) defined PCTs as “designed for the primary purpose of informing decision makers regarding the comparative balance of benefits, burdens, and risks of a biomedical or behavioral health intervention at the individual or population level”.
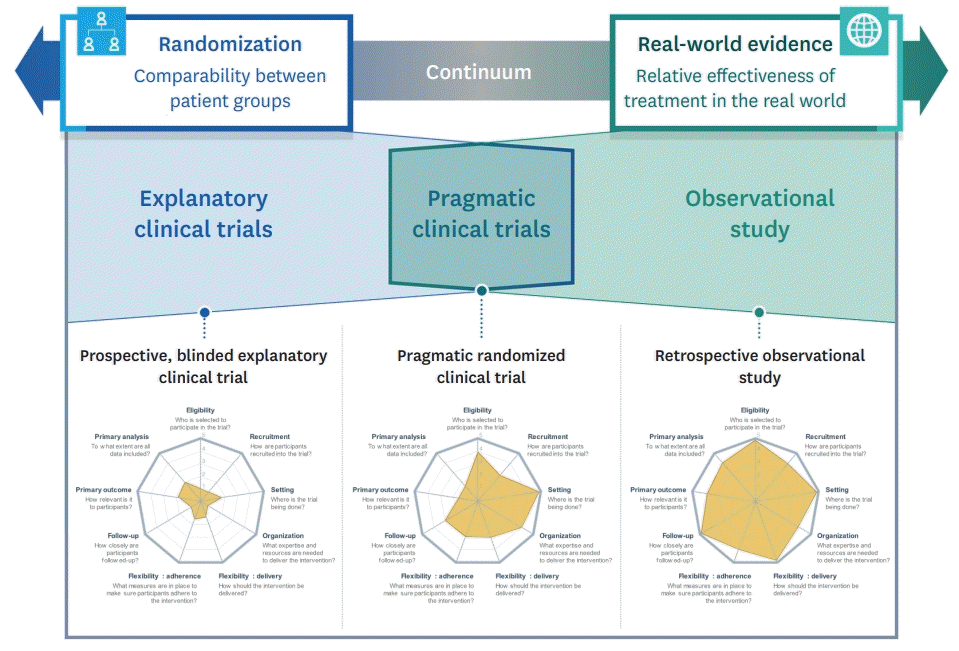 | Figure 1.Relationships among the explanatory clinical trials, observational study, and pragmatic clinical trials, including hypothetical PRECIS-2 diagrams. PRECIS-2 diagrams in the figure is referred to Bevan et al.20)
PRECIS = PRagmatic Explanatory Continuum Indicator Summary.
|
Table 2.
Information in the table is referred to Loudon et al.18)
PRECIS = PRagmatic Explanatory Continuum Indicator Summary.
![]()
STEPS IN CONDUCTING PCTs
Learning what makes PCTs different
Building partnership to ensure a successful trial
Planning for sustainability before launching of PCTs
Choosing the right outcomes
Planning the study design and analysis
Study sites: The sites in which PCTs are performed should be similar to the actual clinical setting and should be representative.12) Sites should be carefully selected because the prevalence of diseases and the availability of medical care may vary greatly depending on the type and region of the institutions.32) In order to select the sites for PCTs, the heterogeneity, patient characteristics, and disease prevalence of the sites should be assessed.32) Then, the feasibility and capacity of the study should be confirmed, including interest in study objectives, level of study experience, and number of patients accessible.32) PCTs should include several heterogeneous sites because if only single or homogeneous sites are included, the test results may not be applicable to patients in various settings.
Randomization: Randomization can help reduce the selection bias caused by the differences in study groups; any difference in the outcomes can be attributed to the assignment of treatments. Therefore, randomization should be applied in RCTs including PCTs.33) Even though randomization at both the patient level and cluster (e.g., sites and physicians) level12) is possible in PCTs, randomizing at the patient level may affect patients to select intervention, which may result in low recruitment rates.34) Therefore, cluster randomization, which involves the assignment of treatments at the level of physicians, hospitals, or other units handling multiple patients, is useful.34) There are several types of cluster randomization: cluster with crossover randomized design, cluster with partial crossover randomized design, and simple cluster-randomized design.35)
Blinding: Blinding of patients and health care providers during the treatment period is useful to minimize bias and enhance the internal validity in most clinical trials such as ECTs. In PCTs, it is generally not appropriate to use placebo and blinding owing to ethical reasons, preference of non-blinded trials by patients, and the specificity of PCTs.16) Therefore, the following methods can be recommended to minimize bias when patients or investigators are not blinded in PCTs12): 1) use of hard endpoints (i.e., objective endpoints) rather than subjective endpoints, 2) determination of endpoints from blinded health care professionals, and 3) blinding of statisticians and data analysts during the trial and planning of analysis, if possible. If blinding is not possible for the individual patients and investigators, the cluster randomization method can be used.36)37) The lack of blinding in PCTs decreases the internal validity, but it will be easier to generalize the test results by increasing the external validity.16)
Statistical analyses: The statistical analysis methods used mainly in PCTs must be fully understood, and an analysis plan should be made. The analysis method of RCTs can be applied to PCTs, but considerations are required because of the uniqueness of PCTs.38) Intention to treat (ITT) analysis and per-protocol (PP) analysis are required in analyzing the results of clinical trials, and ITT is considered the principal analysis in PCTs.39) Although problems of validity may occur if the randomized patients switch to other alternative treatments during the ITT analysis, ITT is generally recommended as it reduces the risk of selection bias and the unguaranteed covariate balance that can occur in PP analysis.38) However, large amounts of missing data in the ITT analysis can cause a confounding bias.38) To solve this, a follow-up history and the possible reasons for missing data should be recorded for all patients, which is the basis for analyzing the extent and the pattern of missing data. Bias because of missing data can be supplemented using causal inference tools such as a marginal structural model or through inclusion of additional data such as PROs from other sources (e.g., wearable devices).12) In addition, PCTs include more heterogeneous populations than RCTs; hence, the responses of subgroups to interventions may differ.38) When evaluating the effectiveness of treatments in subgroup analyses, a priori subgroup analysis with a large sample size should be used to ensure valid estimates of treatment effects and avoid obtaining false positive results.
Considering oversight and monitoring
Testing the feasibility of trials
Preparations for the launching of PCTs
Preparing for dissemination and implementation
EXAMPLES OF PCTs
Table 3.
| Trial | Year | Ref. | Question | Inclusion criteria of participants (No. of participants; No. of sites) | Intervention (No. of participants) vs. Comparator (No. of participants) | Randomization | Study period† (follow-up period) | Primary outcomes | Results |
|---|---|---|---|---|---|---|---|---|---|
| CRASH-2 | 2020 | 56 | Whether the early administration of tranexamic acid reduces death, vascular occlusive events, and the receipt of blood transfusion | Adult trauma patients with significant haemorrhage and who were within 8 hours of injury (n=20,211; n=274) | Tranexamic acid (n=10,060) vs. Placebo (n=10,067) | Central randomization at the level of individuals | May 2005 to Mar 2010 (4 weeks) | Death in hospital | Tranexamic acid safely reduced the risk of death in bleeding trauma patients |
| Post-MI FREEE | 2011 | 61 | Whether full prescription drug coverage for statins, β-blockers, ACE inhibitors, and ARBs is more effective to patients after MI | Adults discharged alive from hospital after MI who received health services and prescription drug benefits through Aetna, Inc. (n=5,855; N/A*) | Full prescription coverage (n=2,845) vs. Usual prescription coverage (n=3,010) | Cluster randomization at the level of the plan sponsor | Nov 2007 to Nov 2010 (≥1 year) | First vascular event or revascularization | The removal of copayments for drugs prescribed after being discharged due to MI did not significantly reduce rates of the primary outcome |
| TASTE | 2013 | 62 | Whether thrombus aspiration before PCI reduces mortality than usual care in patients with STEMI | Patients in a registry for coronary angiography and angioplasty (n=7,244; n=31) | Thrombus aspiration before PCI (n=3,621) vs. Usual care (n=3,623) | Registry based randomization | Jul 2010 to Aug 2013 (30 days) | All-cause death | Thrombus aspiration before PCI did not reduce 30-day mortality in patients with STEMI compared to PCI alone |
| SLS | 2016 | 64 | Whether the effectiveness and safety of the once-daily inhaled combination of fluticasone furoate–vilanterol is better than existing maintenance therapy | Adults (≥40) who received diagnosis of COPD and who had ≥1 COPD exacerbations in the previous 3 years in 75 general practices (n=2,799; n=75) | Fluticasone furoate- vilanterol (n=2,799) vs. Usual care (n=1,403) | Central randomization at the level of individuals | Mar 2012 to Nov 2015 (1 year) | Mean annual rate of exacerbations | Once-daily combined treatment of fluticasone furoate and vilanterol decreased the rate of exacerbations without a further risk of serious adverse events than usual care |
| High-STEACS | 2018 | 66 | Whether the use of hs-cTnI assay reduces MI or cardiovascular death compared with standard troponin assay | Patients with acute coronary syndrome suspects in 10 Scotland hospitals (n=48,282; n=10) | Reclassified as MI by hs-cTnI assay (n=1,771) vs. Classified as MI by cardiac troponin I assay (n=8,589) | Stepped wedge cluster randomization at the level of the hospital site | Jun 2013 to Mar 2017 (1 year) | Subsequent MI or cardiovascular death | High-sensitivity assay did not reduce in MI or cardiovascular death within 1 year |
ACE = angiotensin-converting enzyme; ARBs = angiotensin II receptor blockers; CRASH-2 = Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage; COPD = chronic obstructive pulmonary disease; High-STEACS = High-Sensitivity Troponin in the Evaluation of patients with Acute Coronary Syndrome; hs-cTnI = high-sensitivity cardiac troponin I; MI = myocardial infarction; PCI = percutaneous coronary intervention; PCT = randomized controlled trial; Post-MI FREEE = The Post-Myocardial Infarction Free Rx and Economic Evaluation; SLS = Salford Lung Study; STEMI = ST-segment elevation myocardial infarction; TASTE = Thrombus Aspiration in ST-Elevation Myocardial Infarction in Scandinavia.
![]()
 XML Download
XML Download