

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Autophagy, literally meaning ‘self-devouring,’ is a subcellular process disintegrating cell’s own material such as proteins, lipids and organelles including mitochondria or endoplasmic reticulum (ER) in lysosome for supply of essential nutrients in nutrient deficiency or turnover of dysfunctional or senescent organelle.12 Three major types of autophagy such as macroautophagy, microautophagy and chaperone-mediated autophagy were described. Among them, macroautophagy (henceforth referred to as ‘autophagy’) has been most extensively studied and is characterized by the rearrangement of subcellular membrane which leads to sequestration of cytoplasm, target substances or organelles and formation of double membrane-surrounded structure called ‘autophagosome.’ Autophagosome formation is not the end of autophagic process, and autophagosome is fused to lysosomes, forming autophagolysosome in which lysosomal enzymes degrade sequestered material or organelles.1 Thus, lysosome is an effector organelle in the execution of autophagy. The main function of autophagy is maintenance of intracellular nutrient balance through supply of essential elements in nutrient deficiency and quality control of proteins or organelles through their degradation and rejuvenation. Provision of vital nutrients during energy crisis through autophagy-dependent degradation of intracellular macromolecules appears to be evolved from ancient cellular machinery protecting against nutritional deficiency of unicellular organisms such as yeast, and is critical for mammalian system as well since neonatal mice with knockout (KO) of essential autophagy genes succumb to death during the first several hours after birth, which was ascribed to the inability to supply nutrient to the brain due to inability to suckle during the immediate postnatal period.34
While the phenomenon of autophagy has been known for several decades,567 molecular mechanism of autophagy machinery began to be characterized since the 1990s using yeast system.8 To understand detailed molecular and cellular mechanism of autophagy, readers are encouraged to consult excellent recent review articles.910
Since autophagy is critical in the maintenance of cellular homeostasis and organelle function, autophagy can affect almost all pathophysiological processes, and dysregulated autophagy is likely to be involved in the pathogenesis of diverse diseases including metabolic disorders such as type 2 diabetes (T2D) or metabolic syndrome, neurodegenerative disorders such as Alzheimer’s disease or Parkinson’s disease, inflammatory disorders such as inflammatory bowel disease, infectious diseases, cancer and aging. For treatment of those diseases, therapeutic modulation of autophagy might be a future option.111213 In the case of metabolic diseases and T2D, insulin and its downstream mTORC1 are well-known inhibitors of autophagy,14 while glucagon can activate autophagy.6 Furthermore, function of mitochondria and ER that are closely related to β-cell survival or death and insulin resistance or sensitivity,15161718 is critically dependent on autophagy.1920 Thus, dysregulation of autophagy could be an important mechanism in the pathogenesis of metabolic disorders and T2D,21 which is supported by GWAS studies showing the association between single nucleotide polymorphism of several lysosomal genes and the development of T2D.22
In this review, we summarize the recent findings regarding the role of autophagy or lysosomal function in the pathogenesis of T2D and potential role of modulation of autophagy or lysosomal function as a new therapeutic modality against metabolic disorders.
Go to : 
ROLE OF AUTOPHAGY IN OBESITY-INDUCED METABOLIC CHANGES AND DIABETES
Despite extensive studies by many investigators, the role of autophagy in the development of metabolic disorders is not clearly elucidated, which is partly due to the aforementioned neonatal lethality of mice with global KO of essential autophagy genes.34 Therefore, role of autophagy in metabolic diseases in vivo has been studied largely using site-specific autophagy gene KO mouse models that showed distinct metabolic phenotypes depending on the tissues in which the autophagy gene was knocked out.23
Thus, an important question regarding the role of autophagy in metabolic diseases and T2D is what the role of systemic autophagy insufficiency of physiological range in the development of metabolic disorders is. This question was partially addressed in a previous study using mice with Atg7 heterozygote or haploinsufficient mice.24
Atg7+/- heterozygote mice showed no apparent metabolic abnormality when fed normal chow diet imposing no metabolic stress. In contrast, when fed high-fat diet (HFD) or bred with ob/w mice to impose metabolic stress, mice with autophagy haploinsufficiency displayed more severe and persistent diabetes compared to autophagy-competent counterparts, accompanied by aggravated insulin resistance and glucose intolerance (Fig. 1A and B). Microscopic examination of metabolic tissues showed more severe accumulation of lipids in hepatic sections, indicating aggravated fatty liver (Fig. 1C). In adipose tissue, increased number of crown-like structure (dead adipocytes surrounded by macrophages representing metabolic inflammation) was observed, suggesting increased metabolic inflammation. Elevated levels of serum alanine aminotransferase and aspartate aminotransferase suggesting augmented liver injury due to aggravated fatty liver and a higher expression of inflammatory cytokines or chemokines in adipose tissue were also observed. Aggravated fatty liver of Atg7+/--ob/ob mice could be due to reduced lipophagy in hepatocytes. Mechanism of augmented metabolic inflammation was due to stronger activation of inflammasome, accompanied by increased cleavage of pro-caspase-1 and maturation of pro-IL-1β into IL-1β in adipose tissue macrophages (ATMs) of Atg7+/--ob/ob mice compared to autophagy-competent control mice (Fig. 1D).
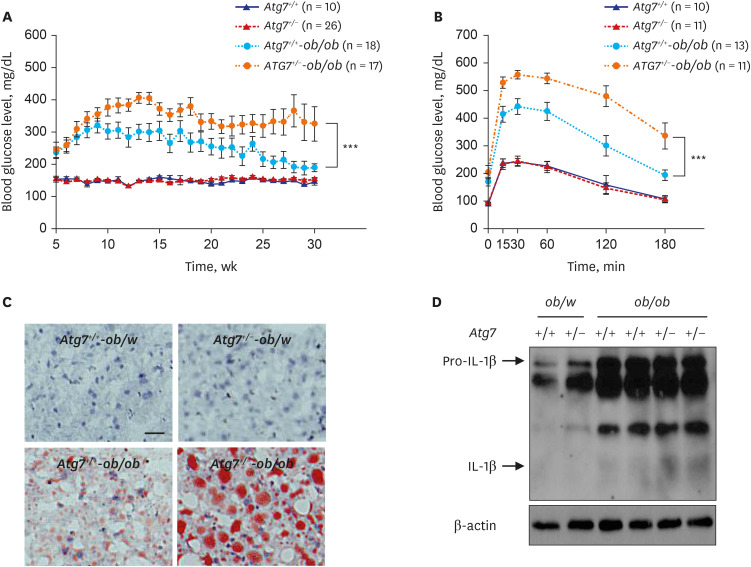 | Fig. 1Aggravated metabolic profile and inflammasome activation in obese mice with autophagy haploinsufficiency. (A) Monitoring of non-fasting blood glucose level showed more severe and persistent diabetes in autophagy-haploinsufficient Atg7+/--ob/ob mice compared to autophagy-competent Atg7+/+-ob/ob mice. (B) Intraperitoneal glucose tolerance test showed more severe glucose intolerance in Atg7+/--ob/ob mice compared to Atg7+/+-ob/ob mice. (C) Lipid content in the liver tissue assessed by ORO staining was higher in Atg7+/--ob/ob mice compared to Atg7+/+-ob/ob mice, showing aggravated fatty liver. (D) Immunoblot showed increased maturation of pro-IL-1β to IL-1β in stromal vascular fraction of adipose tissue from Atg7+/--ob/ob mice compared to that from Atg7+/+-ob/ob mice.IL = interleukin.
Reproduced from Lim et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun 2014;5:4934.
|
Increased infiltration of macrophages expressing IL-1β into the adipose tissue of Atg7+/--ob/ob mice was also observed by confocal microscopy. Augmented inflammasome activation in ATMs of Atg7+/--ob/ob mice was likely due to aggravated mitochondrial dysfunction in metabolically-stressed macrophages, which plays a critical role in inflammasome activation by NACHT, LRR and PYD domains-containing protein 3 (NLRP3) activators.24252627 Indeed, accumulation of mitochondrial reactive oxygen species and the proportion of cells with lower mitochondrial potential after treatment with palmitic acid in conjunction with lipopolysaccharide (LPS) were augmented in Atg7+/- macrophages compared to autophagy-competent macrophages, likely due to compromised removal of dysfunctional mitochondria in autophagy-insufficient macrophages.
These results suggest that increased lipid content in metabolic tissue likely due to insufficient lipophagy and augmented inflammasome activation due to insufficient removal of dysfunctional mitochondria acting as a hub for inflammasome activation25 are main culprits leading to further aggravated metabolic profile of Atg7+/--ob/ob mice compared to autophagy-competent counterpart. In fact, these two events are interrelated because certain fatty acids, effector molecules of lipid injury, can impose mitochondrial stress2829 and also activate NLRP3.2627 Since tissue lipid accumulation can cause ER stress,30 autophagy insufficiency can aggravate both ER stress and mitochondrial stress associated with lipid injury, two major stress associated with the development of T2D in lipid overload, obesity or metabolically stressed state, which is consistent with the concept that autophagy can rejuvenate stressed organelles such as ER and mitochondria. These results suggest that autophagy insufficiency of physiological range might not cause significant trouble in the basal metabolic state but can lead to deleterious events when metabolic stress is imposed. This suggestion is supported by the findings of previous papers showing Atg4b-KO mice with a partial disruption of the autophagic process showed more severe fatty liver, glucose intolerance, insulin resistance and weight gain compared to control mice after feeding hypercaloric diets.31 In a similar vein, HFD-fed mice with defective stimulus-induced autophagy due to knock-in mutations of BCL2 phosphorylation sites (Thr69Ala, Ser70Ala and Ser84Ala) showed impaired exercise-induced metabolic improvement.32 On the other hand, Atg5 overexpression has been shown to improve metabolic deterioration associated with aging,33 suggesting the possibility that enhancement of autophagy could be a therapeutic option against metabolic deterioration due to aging or other causes.
Go to :
AUTOPHAGY MODULATORS IN OBESITY-INDUCED METABOLIC CHANGES AND DIABETES
These results suggested the possibility that autophagy insufficiency due to aging, genetic causes and environmental or exogenous factors could be an underlying cause of T2D. Autophagy insufficiency would impair rejuvenation of stressed or dysfunctional ER or mitochondria which could play an important role in the development of T2D15161718 when metabolic stress is imposed. Conversely, these data suggest the possibility that administration of pharmacological agents enhancing autophagic activity may have beneficial metabolic effects in metabolically stressed conditions such as obesity or aging. Several previous studies have attempted to enhance autophagic activity using imatinib or trehalose, which showed improvement of metabolic profile of metabolically stressed experimental animals.34353637 In such studies showing metabolic improvement by putative autophagy enhancers, reduced inflammasome activation and metabolic inflammation in metabolic tissues were observed.24 Several known anti-diabetic drugs such as metformin, glucagon-like peptide 1 (GLP-1) receptor agonists and PPAR agonists have also been claimed to exert their therapeutic effects through enhancement of autophagic activity11 which might be related to TFEB activation.3839 These results suggest the possibility that if authentic autophagy enhancers without side effects could be developed, they could be candidates for therapeutic agents of novel concept against diabetes, metabolic syndrome associated with lipid overload or cardiovascular diseases.
We recently identified and developed autophagy enhancer small molecules in a screening of a chemical library employing Renilla luciferase conjugated to LC3.40 Using this system, we were able to identify several chemicals that induce autophagic activity to the lysosomal degradation steps of autophagy and, thus, are activators of autophagic flux rather than autophagy level.41 Among candidate chemicals that could enhance degradation of LC3-conjugated Renilla luciferase, we chose MSL that did not inhibit mTORC1 since mTORC1 inhibitors such as rapamycin are well-known autophagy enhancers but can cause unwanted metabolic adverse effects such as reduction of pancreatic β-cell mass or augmented insulin resistance.4243 To further investigate the mechanism of mTORC1-independent autophagy induction, we found that MSL can enhance nuclear translocation of TFEB, a master regulator of autophagy gene expression and lysosomal biogenesis.44 We also observed that MSL does not increase lysosomal Ca2+ efflux but induce dephosphorylation of TFEB by enhancing calcineurin activity. We chemically modified MSL to increase microsomal stability (MSL-7), and MSL-7 was able to exert beneficial metabolic effects in ob/ob or HFD-fed mouse models, as shown by reduction of non-fasting blood glucose level and amelioration of glucose intolerance (Fig. 2A and B). In metabolic tissues, MSL-7 reduced fatty liver probably by enhancing the clearance of lipid in the hepatic tissue and ameliorated metabolic inflammation in adipose tissue (Figs. 2C, 3A and B).41
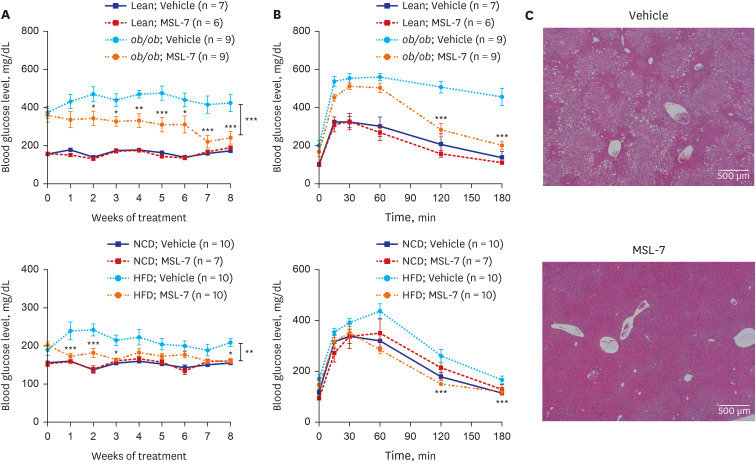 | Fig. 2Improved glucose profile and fatty liver of obese mice by autophagy enhancer. (A) Monitoring of non-fasting blood glucose levels showed improved glucose profile after administration of MSL-7, an autophagy enhancer, to ob/ob (upper) and HFD-fed mice (lower). (B) Intraperitoneal glucose tolerance test showed improved glucose tolerance after administration of MSL-7 to ob/ob (upper) and HFD-fed mice (lower). (C) Hematoxyling and eosin staining showed ameliorated fatty liver after administration of MSL-7 to ob/ob mice.HFD = high-fat diet.
Reproduced from Lim et al. A novel autophagy enhancer as therapeutic agent against metabolic syndrome and diabetes. Nat Commun 2018;9:1483.
|
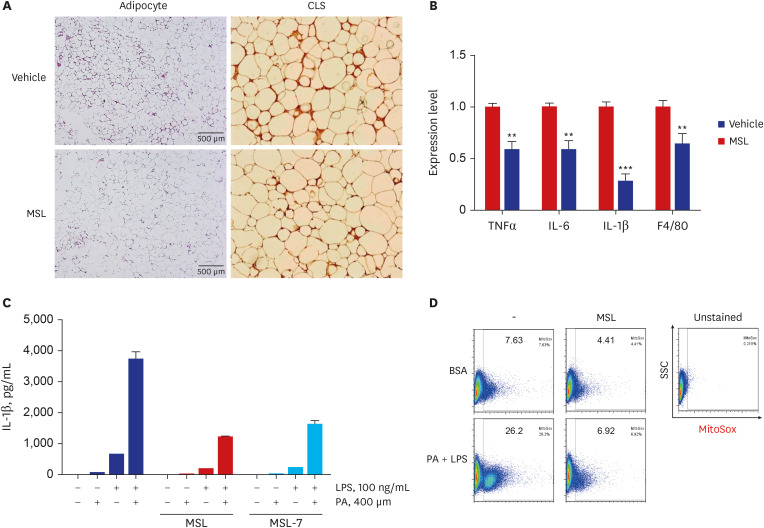 | Fig. 3Reduced metabolic inflammation and inflammasome activation by autophagy enhancer. (A) Immunohistochemistry of adipose tissue sections from ob/ob mice using F4/80 antibody showed decreased number of CLS after administration of MSL for 8 weeks. (B) Real-time reverse transcription polymerase chain reaction using mRNA from adipose tissue showed reduced expression of inflammatory cytokines after administration of MSL for 8 weeks. (C) Content of IL-1β in the culture supernatant of primary peritoneal macrophages treated with 200 mM PA and/or 500 ng/mL LPS was reduced by MSL or MSL-7, as determined by enzyme-linked immunosorbent assay. (D) Mitochondrial ROS in primary peritoneal macrophages treated with 200 mM PA and/or 500 ng/mL LPS was reduced by MSL, as determined by MitoSox Red staining followed by FACS analysis.CLS = crown-like structure, TNF = tumor necrosis factor, IL = interleukin, PA = palmitic acid, LPS = lipopolysaccharide.
Reproduced from Lim H et al. A novel autophagy enhancer as therapeutic agent against metabolic syndrome and diabetes. Nat Commun 9:1483, 2018
|
Alleviation of metabolic inflammation was due to attenuation of fatty acid-induced mitochondrial stress or dysfunction and subsequent downregulation of fatty acid-induced inflammasome activation by autophagy enhancer since dysfunctional mitochondria is an important player in inflammasome activation (Fig. 3B-D).25 These results suggest that autophagy enhancer small molecules can improve metabolic profile of obese mice by improving lipid clearance and attenuating metabolic inflammation through rejuvenation of mitochondrial function (Fig. 4).
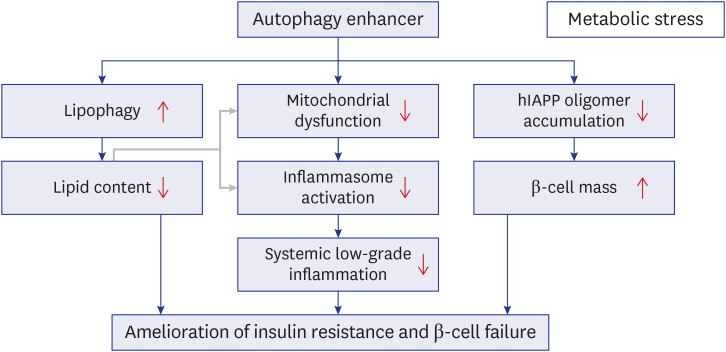 | Fig. 4Proposed model for improved metabolic profile in obesity-induced diabetes and human-type diabetes by an autophagy enhancer (MSL-7). Autophagy enhancer can reduce tissue lipid accumulation through activation of lipophagy in the presence of metabolic stress or lipid overload (left axis). Autophagy enhancer can also expedite rejuvenation of stressed or dysfunctional mitochondria which plays an important role in inflammasome activation, and, thereby, reduce metabolic inflammation (middle axis). Lipids such as palmitic acid can induce mitochondrial stress and activate inflammasome (gray arrow). Autophagy enhancer can accelerate clearance of hIAPP oligomer in pancreatic β-cells and protect β-cells from hIAPP oligomer-mediated apoptosis since hIAPP clearance is predominantly cleared by autophagy or lysosomal degradation rather than proteasomal degradation (right axis). Combined effects of these three axes and their interaction finally lead to the improvement of metabolic profile of obesity-induced diabetes and human-type diabetes characterized by islet amyloid accumulation through amelioration of insulin resistance and improvement of β-cell function.hIAPP = human islet amyloid polypeptide.
|
Go to :
ROLF OF AUTOPHAGY IN ISLET AMYLOID-INDUCED DIABETES
In the previous sections, we discussed the role of autophagy in diabetes and potential therapeutic modulation of autophagy based on data from animal models of T2D and metabolic syndrome. However, human diabetes and murine one differ in several aspects. One of the key differences is islet amyloid deposition in human diabetes but not in murine diabetes. Islet amyloid stained with Congo Red is found in over 90% of human patients with T2D.45 This difference is due to non-identical amino acid sequence of islet amyloid polypeptide (IAPP) (previously called amylin). Human IAPP (hIAPP) is amyloidogenic, while murine IAPP (mIAPP) is non-amyloidogenic due to proline-rich sequences in aa 26-29 that inhibits oligomerization or amyloid formation.46 It is an open question why human IAPP acquired amyloidogenicity during evolution, while physiological function of IAPP has been ascribed to regulation of insulin secretion, protection of β-cells and tumor suppression.4748 Then, amyloidogenic or aggregate proteins are preferentially cleared by autophagy or lysosomal proteolysis, while usual non-amyloidogenic soluble proteins are cleared by both proteasomal and autophagic degradation pathways.49 Thus, the role of autophagy in human diabetes could be larger than that in murine one. As mice do not express amyloidogenic IAPP, transgenic mice expressing hIAPP specifically in pancreatic β-cells were employed to study the role of autophagy in human-type diabetes characterized by islet amyloid deposition, (hIAPP+ mice).5051
hIAPP+ mice develop mild hyperglycemia but not diabetes. However, after crossing to autophagy-deficient mice, hIAPP+ mice with β-cell-specific autophagy deficiency (hIhIAPP+
Atg7Δb-cell mice) developed severe diabetes.5253 These data underscore the role of β-cell autophagy in the abrogation of hIAPP-induced diabetes without which diabetes occurs, probably by clearing hIAPP oligomer and islet amyloid. The importance of hIAPP and its clearance in the pathogenesis of human-type diabetes were also investigated using hIAPP knock-in mice that expresses more physiological level of hIAPP compared to hIAPP+ mice. In line with the results of hIAPP+
Atg7Δb-cell mice, HFD-fed hIAPP knock-in:Atg7Δβ-cell mice developed more severe metabolic impairment and further reduction of β-cell mass compared to HFD-fed Atg7Δβ-cell mice with wild-type endogenous mIAPP.54 This result suggests that hIAPP oligomer of physiological level is cleared by autophagy without which metabolic impairment ensues in the presence of metabolic stress and underscore the role of appropriate β-cell autophagy in the protection against human diabetes in the presence of metabolic stress.
Regarding the mechanism of diabetes development in hIAPP+
Atg7Δb-cell mice, hIAPP oligomer stained with oligomer-specific antibody (I11 antibody raised against hIAPP oligomer) and islet amyloid stained with EE-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (FSB) or Thioflavin-S, accumulated in pancreatic islets of hIAPP+
Atg7Δb-cell mice.52 Accumulation of hIAPP oligomer can be an important event leading to β-cell injury and diabetes of hIAPP+
Atg7Δb-cell mice, as hIAPP oligomer rather than hIAPP islet amyloid could be an effector molecule imposing β-cell death, as suggested by the ‘toxic oligomer hypothesis.’55 Consistently, increased apoptosis of pancreatic β-cells stained with TUNEL reagent was observed in the pancreatic islets of hIAPP+
Atg7Δb-cell mice compared to hIAPP+
Atg7F/F or hIAPP-
Atg7Δb-cell mice. The types of hIAPP oligomers in autophagy-deficient β-cells were also investigated. To this end, HA sequence was attached to prepro-hIAPP and -mIAPP. When INS-1 insulinoma cells were transfected with prepro-mIAPP, only pro-mIAPP monomer after cleavage of ‘pre’ sequence was observed as revealed by immunoblot analysis using anti-HA antibody. In contrast, when prepro-hIAPP was expressed, pro-hIAPP dimer was observed in addition to pro-hIAPP monomer. When autophagy was inhibited with 3-methyladenine (3-MA), accumulation of pro-hIAPP dimer was increased and a protein band consistent with pro-hIAPP trimer was also observed. Accumulation of pro-hIAPP dimer and trimer was more prominent in detergent-insoluble fraction compared to detergent-soluble fraction, suggesting that pro-hIAPP dimer might be formed initially in membrane-rich compartment and translocate to the soluble fraction or proceed to pro-hIAPP trimer in the membrane fraction. Accumulation of pro-hIAPP dimer or trimer disappeared when three amino acids in aa 26–29 were changed to prolines that are seen in non-amyloidogenic pro-mIAPP. These results demonstrate that amino acid sequence in the critical region determines oligomerization propensity or amyloidogenicity of IAPP and also suggest the possibility that pro-hIAPP dimer or trimer could be the initial nidus for further progression to high-n hIAPP oligomer and finally hIAPP amyloid. These results are consistent with a previous paper showing that pro-hIAPP aggregates are dominant forms in the initial phase of islet amyloid formation which may act as seeds for further deposition of mature IAPP in the later phase, finally leading to the formation of intracellular and extracellular amyloid.56 Biochemical reason for preferential role of pro-hIAPP dimer or trimer rather than mature hIAPP dimer or trimer in the initial phase of islet amyloid formation could be due to a heparin binding domain located in the N-terminal cleavage site and a higher isoelectric point of pro-hIAPP compared to mature hIAPP favoring pro-hIAPP association with negative-charged membranes.565758
The role of endogenous IAPP oligomer has also been studied instead of transgenic hIAPP oligomer. When monkey islet cells expressing amyloidogenic simian IAPP (sIAPP) with only one amino acid sequence difference from hIAPP sequence were incubated with bafilomycin A blocking lysosomal step of autophagy, accumulation of intracellular sIAPP oligomer accompanied by apoptosis occurred in vitro,52 suggesting the role of autophagy in the clearance of endogenous sIAPP oligomer, which was similar to that of genetically expressed hIAPP oligomer.
Go to :
AUTOPHAGY MODULATORS IN ISLET AMYLOID-INDUCED DIABETES
As autophagy deficiency can lead to augmented hIAPP oligomer accumulation or β-cell apoptosis and also diabetes, the converse question - whether autophagy enhancement improve β-cell function by accelerating clearance of hIAPP oligomer and inhibiting hIAPP oligomer-induced β-cell apoptosis - was tested. To this end, trehalose has been administered to hIAPP+ mice rendered diabetic by feeding HFD. After trehalose administration for 4 weeks, glucose profile of HFD-fed hIAPP+ mice was improved, accompanied by reduced hIAPP oligomer or islet amyloid accumulation, ameliorated β-cell death and improved insulinogenic index representing enhanced β-cell function,52 suggesting possible effect of therapeutic autophagy enhancement in the treatment of human diabetes.
We also studied the effect of aforementioned autophagy enhancer MSL-7 on diabetes of hIAPP+ mice fed HFD, since amyloidogenic or aggregate proteins are preferentially cleared by autophagy or lysosomal proteolysis rather than proteasomal degradation.49 We first tested whether MSL-7 could improve clearance of pro-hIAPP dimer mentioned above. As discussed, pro-hIAPP dimer was observed after transfection of prepro-hIAPP which was increased in the presence of 3-MA increasing pro-hIAPP dimer accumulation. pro-IAPP dimer accumulation was not observed after that of prepro-mIAPP. In this condition, MSL-7 attenuated accumulation of pro-hIAPP dimer in the presence of 3-MA. This effect of MSL-7 was abrogated by bafilomycin A blocking lysosomal proteolysis. Bafilomycin A treatment induced accumulation of pro-hIAPP trimer as well. Probably because of pro-hIAPP oligomer accumulation including pro-hIAPP dimer, apoptosis determined by measuring oligonucleosomal DNA content in the cell lysate was observed after transfection of prepro-hIAPP in the presence of 3-MA but not after that of prepro-mIAPP. MSL-7 attenuated apoptosis of INS-1 insulinoma cells observed after transfection with prepro-hIAPP in the presence of 3-MA, likely due to diminution of pro-hIAPP oligomer accumulation including pro-hIAPP dimer. Importantly, reduced accumulation of pro-hIAPP dimer and ameliorated INS-1 insulinoma cell apoptosis after prepro-hIAPP transfection by MSL-7 were significantly less pronounced in Tfeb-KO INS-1 cells generated by CRISPR/Cas9 technology, indicating the role of Tfeb and Tfeb-mediated autophagy in the effects of MSL-7.59
We also employed endogenous system instead of transfection system using amyloidogenic prepro-hIAPP in the next experiment to study the role of autophagy enhancers on hIAPP oligomer clearance. When primary monkey islet cells expressing amyloidogenic sIAPP were prepared and incubated with 3-MA, accumulation of sIAPP oligomer stained with A11 antibody was observed. A11 antibody has been raised against Aβ oligomer but recognizes hIAPP oligomer, showing the similarity between conformation of Aβ oligomer and that of hIAPP oligomer, despite no similarity in the amino acid sequences between them.6061 sIAPP oligomer accumulating in the presence of 3-MA was diminished by MSL-7,59 suggesting that MSL-7 can clear not only pro-hIAPP oligomer in cells transfected with amyloidogenic prepro-hIAPP but also endogenous amyloidogenic sIAPP. This effect of MSL-7 was accompanied by TFEB nuclear translocation in monkey islet cells, suggesting that TFEB-mediated induction of autophagy and lysosomal proteolysis is responsible for the effects of MSL-7. Further, colocalization between sIAPP oligomer and LC3 was observed in the presence of bafilomycin A that inhibits disappearance of sIAPP oligomer, suggesting that sIAPP oligomer is cleared by MSL-7-induced autophagy. Likely due to clearance of sIAPP oligomer, MSL-7 reduced apoptosis of monkey islets cells in the presence of 3-MA increasing sIAPP oligomer accumulation. The effect of MSL-7 on the clearance of endogenous hIAPP oligomer was also studied. To this end, β-cells derived by differentiation of induced pluripotent stem (iPS) cells were employed. We confirmed that MSL-7 induced TFEB nuclear translocation in iPS-derived β-cells. When iPS-derived β-cells were incubated with 3-MA, hIAPP oligomer recognized by A11 antibody was observed. In this condition, MSL-7 reduced hIAPP oligomer accumulation in iPS-derived β-cells. Furthermore, MSL-7 reduced the number of TUNEL+ apoptotic cells among iPS cell-derived β-cells in the presence of 3-MA.59
Since these results suggested that MSL-7 can protect β-cells from hIAPP oligomer-induced cell death in vitro, we studied whether MSL-7 can protect β-cells from hIAPP oligomer-induced cell death in vivo. When we administered MSL-7 to hIAPP+ mice for 8 weeks during HFD feeding, MSL-7 was found to improve the non-fasting or fasting blood glucose levels and glucose tolerance of hIAPP+ mice rendered diabetic by HFD feeding, together with enhanced β-cell function represented by restored insulinogenic index59 (Fig. 5A-D). Reduced accumulation of hIAPP oligomer or islet amyloid, amelioration of β-cell apoptosis and restoration of β-cell mass were also observed (Fig. 6A-D). Beneficial effects of MSL-7 on β-cell viability and function was accompanied by restoration of diminished mitochondrial respiration such as basal, glucose-stimulated, ATP-coupled and maximal oxygen consumption of primary β-cells from hIAPP+ mice fed HFD. Importantly, improvement in β-cell function represented by insulinogenic index and glucose profile by MSL-7 was significantly diminished by β-cell-specific Tfeb-KO, suggesting that the in vivo effects of MSL-7 are dependent on TFEB activation. Reduced accumulation of hIAPP oligomer, islet amyloid and amelioration of β-cell apoptosis by MSL-7 were also significantly less pronounced in Tfeb-KO hIAPP+ mice fed HFD compared to control mice. Intriguingly, glucose profile and insulinogenic index representing β-cell function were impaired in Tfeb-KO hIAPP+ mice fed HFD compared to hIAPP+ mice with wild-type Tfeb fed HFD mice even before administration of MSL-7, suggesting that bulk autophagy or other types of selective autophagy that is dependent on Tfeb could play a role in the maintenance or adaptation of pancreatic β-cells to metabolic stress.59
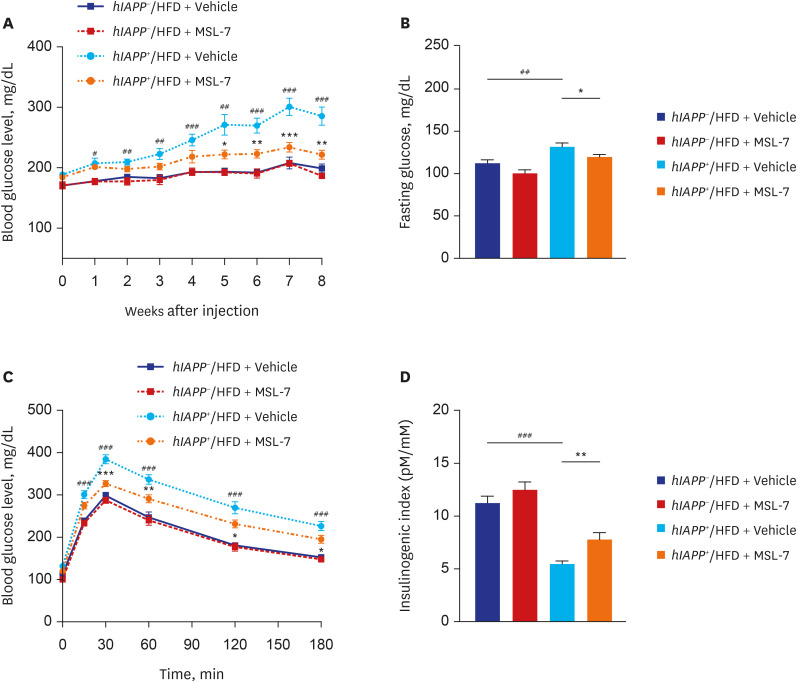 | Fig. 5Metabolic effect of autophagy enhancer on human-type diabetes of hIAPP+ mice. (A) Monitoring of non-fasting blood glucose in hIAPP+ mice during HFD feeding showed amelioration of diabetes in HFD-fed hIAPP+ mice by MSL-7, an autophagy enhancer. (B) Fasting blood glucose of HFD-fed hIAPP+ mice was also reduced by MSL-7 treatment for 8 weeks. (C) Intraperitoneal glucose tolerance test showed improved glucose tolerance by MSL-7 administration to HFD-fed hIAPP+ mice for 8 weeks. (D) Insulinogenic index representing β-cell function was also improved by MSL-7 administration to HFD-fed hIAPP+ mice for 8 weeks.hIAPP = human islet amyloid polypeptide, HFD = high-fat diet.
Reproduced from Kim et al. An autophagy enhancer ameliorates diabetes of human IAPP-transgenic mice through clearance of amyloidogenic oligomer. Nat Commun 2021;12:183..
|
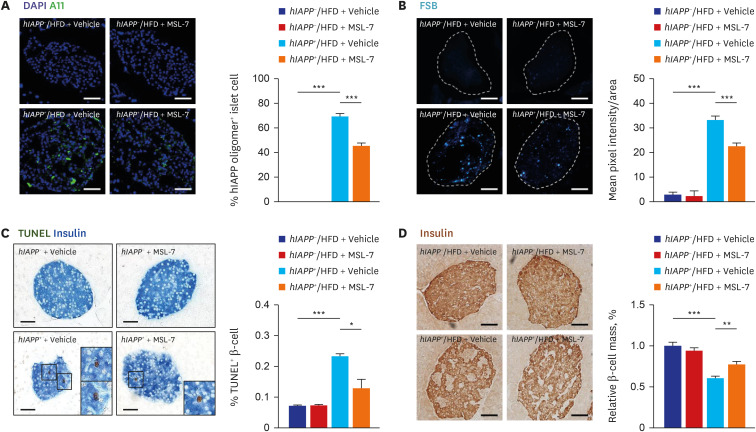 | Fig. 6Pancreatic islets of human-type diabetes of hIAPP+ mice treated with autophagy enhancer. (A) Immunostaining with A11 antibody showed that accumulation of hIAPP oligomer in pancreatic islets of HFD-fed hIAPP+ mice was significantly reduced by treatment with MSL-7, an autophagy enhancer for 8 weeks. (B) FSB staining showed that accumulation of islet amyloid in pancreatic islets of HFD-fed hIAPP+ mice was significantly reduced by treatment with MSL-7 for 8 weeks. (C) TUNEL staining combined with insulin immunohistochemistry showed that the number of apoptotic β-cells in pancreatic islets of HFD-fed hIAPP+ mice was significantly reduced by treatment with MSL-7 for 8 weeks. (D) Relative β-cell mass estimated by insulin immunohistochemistry and subsequent point counting was reduced in HFD-fed hIAPP+ mice, which was significantly improved by MSL-7 treatment for 8 weeks.hIAPP = human islet amyloid polypeptide, HFD = high-fat diet.
Reproduced from Kim et al. An autophagy enhancer ameliorates diabetes of human IAPP-transgenic mice through clearance of amyloidogenic oligomer. Nat Commun 2021;12:183.
|
These results suggest that autophagy enhancer could be a novel therapeutic agent not only against murine diabetes with lipid overload but also against human-type diabetes characterized by islet amyloid accumulation and also the possibility that autophagy enhancer may have more significant effect on human diabetes compared to its effect on murine diabetes because of additional effect of autophagy enhancer on β-cell viability and function (Fig. 4).
Go to :
ROLE OF AUTOPHAGY AND ITS MODULATION IN DIABETES ASSOCIATED WITH MISFOLDED (PRO)INSULIN
Akita mice develop a special type of ER stress-induced diabetes which is due to mutation of 7th cysteine of Ins2 gene, leading to aggregation of misfolded proinsulin. A recent paper reported that removal of Akita aggregates is dependent on autophagy mediated by Beclin 1, Ulk2 and Atg5. Intriguingly, ER-phagy, a selective autophagy rejuvenating dysfunctional or stressed ER, was involved in the autophagic degradation of Akita aggregates. Among several ER-phagy receptors, RTN3, an ER-phagy receptor responsible for the selective degradation of ER tubules (but not other ER-phagy receptors such as FAM134B, Sec62 or CCPG1) was involved,62 suggesting that involvement of a selective type of autophagy in the removal of misfolded proinsulin molecules. Since mutant insulin gene-induced diabetes of young (MIDY) is a special form of human diabetes due to mutations of INS gene, ER-phagy may be important in the clearance of misfolded proinsulin in MIDY, which could have relevance in the treatment of human MIDY patients. While MIDY is a rare form of genetic diabetes, proinsulin misfolding was observed even in normal pancreatic β-cells which was aggravated by obesity, suggesting that accumulation of such misfolded proinsulin could be an early event not only in MIDY but also in common T2D.63
Regarding therapeutic potential of autophagy modulator in ER stress-induced diabetes, a previous paper showed that treatment of Akita mice with rapamycin or Torin1 could increase viability or insulin content of β-cells and improve glucose profile,64 suggesting a possible therapeutic role of autophagy enhancer in the treatment of diabetes characterized by accumulation of misfolded proinsulin. However, potential adverse effects of rapamycin such as decreased β-cell mass or aggravated insulin resistance due to the inhibition of mTORC2 acting as PDK2 for Akt stimulation656667 should be considered.
Go to :
CONCLUSION AND FUTURE PROSPECTS
The above discussion suggests that insufficiency of autophagy or lysosomal proteolysis due to aging, genetic predisposition or environmental factors could be an underlying cause of T2D and metabolic syndrome in that dysfunctional or stressed ER and mitochondria, crucial elements in the pathogenesis of T2D15161718 cannot be managed appropriately in such conditions. Besides bulk autophagy, selective autophagy such as mitophagy or ER-phagy also appears to be involved in the proper function of β-cells626869 and insulin sensitivity.7071 Furthermore, autophagy modulators could have a therapeutic role in the treatment of diabetes characterized by obesity, lipid overload or islet amyloid accumulation. The effect of currently available anti-diabetic drugs such as metformin and GLP-1 receptor agonists could be partly attributable to autophagy activation.
If new autophagy enhancers without adverse effects could be developed, such drugs could be a novel class of anti-diabetic drugs addressing underlying pathogenic mechanism of diabetes. Such autophagy enhancers could also have effects against nonalcoholic steatohepatitis (NASH), a disease related to metabolic syndrome for which no FDA-approved drugs are available. In addition, autophagy enhancers might be effective against Alzheimer’s disease characterized by accumulation of Aβ oligomer and Tau, since clearance mechanism of hIAPP oligomer and that of Aβ oligomer might share similar components of autophagy machinery.
While autophagy enhancers could have potential therapeutic effects against diverse diseases such as metabolic diseases and neurodegenerative diseases, potential risk of ’autophagic cell death’ needs to be considered. Lysosomal dysfunction exists in diverse conditions such as aging, neurodegenerative disorders, cardiac diseases, obesity and kidney diseases.287273747576 Lysosomal dysfunction may be accompanied by accumulation of autophagic intermediates. If excessive pharmacological activation of the proximal steps of autophagy alone occurs in the presence of lysosomal dysfunction, further accumulation of autophagic intermediate may ensue, which may lead to ‘autophagic cell death’ or cell death due to autophagy,287273747576 which may necessitate attention before development and administration of autophagy enhancers.
Go to :
 XML Download
XML Download