

 PDF
PDF Citation
Citation Print
Print




INTRODUCTION
Muscular atrophy is the loss of skeletal muscle mass as a consequence of increased myofibrillar protein degradation and decreased protein synthesis [1]. Muscle atrophy occurs under a variety of conditions, and can lead to serious social debility and associated social costs especially in the recent aging society [2]. Delta-like 1 homolog (DLK1) is a transmembrane protein belonging to the epidermal-growth-factor-like-repeat-containing family and essential for proper skeletal muscle development and regeneration [3]. In addition, treatment with soluble DLK1 peptide attenuates hepatic steatosis and hyperglycemia in vivo [4], implying pleiotropic effects of DLK1 including metabolic regulation. The effects of DLK1 on muscle and other organs can be studied by injection of soluble DLK1 peptide, which is commercially available as a construct of the N-terminal extra cellular domain of DLK1 fused with a human Fc fragment, into animal models. However, the therapeutic effects of DLK1 in muscle atrophy and the underlying mechanisms of the condition have not previously been studied in vivo.
We used dexamethasone (Dex) to establish an experimental model of muscle atrophy [5], as Dex induces muscle wasting and degeneration by stimulating the production of myostatin [6,7]. Myostatin negatively regulates muscle mass and accelerates muscular atrophy during Dex treatment [8]. The ubiquitin-proteasome system plays a major role in the catabolic action of glucocorticoids, and myostatin increases the activity of important muscle-specific ubiquitin E3 ligases, muscle-specific RING finger protein 1 (MuRF1) and atrogin1 [8]. We also used the cardiotoxin (CTX) isolated from Naja pallida, an amphiphilic peptide that damages muscle due to increased production of reactive oxygen species (ROS) secondary to cytosolic calcium overload [9], to induce muscle atrophy in vivo.
In the present study, we investigated whether DLK1 inhibits or attenuates muscular atrophy in Dex- or CTX-induced muscular atrophy models in vivo and in vitro. To assess the effects of DLK1 on muscle quantity and quality, we evaluated morphological changes in muscle fibers and mitochondria of muscle cells, and changes in the expression of myostatin and other markers of muscle atrophy and differentiation.
METHODS
Development of soluble DLK1 protein
A soluble form of DLK1 protein tagged with FLAG (FLAG-DLK1) was provided by Dr. Bum-Chan Park (Y-Biologics, Inc., Daejeon, Korea) and was produced as described previously [4].
Animals and study design
Eight-week-old male C57BL/6J mice were purchased (Joong-ang Experimental Animal Co., Seoul, Korea) and acclimatized for 2 weeks. The mice were housed in an animal facility maintained at 23°C±2°C and 55%±5% humidity. Mice were exposed to a 12 hours/12 hours light/dark cycle and fed an unrestricted standard chow diet. All animals had free access to drinking water, and body weight was measured every 2 days. All mice were fasted for 6 hours before sacrifice. In all cases, mice were anesthetized and sacrificed 24 hours after the final administration of drugs. After mice were sacrificed, blood was collected by cardiac puncture and tibialis anterior (TA) muscle tissue was harvested. Specimens were snap-frozen in liquid nitrogen and maintained at −80°C until analysis. All animal experiments were approved by the Institutional Animal Care and Use Committee of Yonsei University Health System (YUHS-IACUC, 2017−0066, 2018−0194) and complied with the regulations and guidelines of the Animal Protection Act (2008), the Laboratory Animal Act (2008), and the Eighth Edition of the Guide for the Care and Use of Laboratory Animals of NRC (2011).
Mouse model of dexamethasone-induced muscle atrophy
In experiments employing the Dex-induced muscle atrophy model, 10-week-old C57BL/6J male mice were divided into three groups: (1) control group (distilled water [DW] oral gavage and intraperitoneal [IP] phosphate buffered saline [PBS]; n=6); (2) Dex group (Dex oral gavage and IP PBS; n=8), and (3) Dex+DLK1 group (Dex oral gavage and IP DLK1; n=8). Briefly, 1 mg/kg Dex (D2915, Sigma-Aldrich, St. Louis, MO, USA) or DW was administrated by oral gavage once daily for 2 weeks, and DLK1 (0.8 mg/kg/day) or PBS (DaiHanPharm, Ansan, Korea) was administered by IP injection once daily for 2 weeks.
Mouse model of cardiotoxin-induced muscle atrophy
In experiments employing the CTX muscle-crush injury model, 10-week-old mice were divided into three groups: (1) control group (intramuscular [IM] PBS and IP PBS; n=3); (2) CTX group (IM CTX and IP PBS; n=4); and (3) CTX+DLK1 group (IM CTX and IP DLK1; n=3). Briefly, mice were anesthetized, and then CTX (150 μL at 10 μM) isolated from Naja pallida (Latoxan L81-02) was injected into the TA muscle. Beginning the day of IM CTX or PBS injection, DLK1 (0.8 mg/kg/day) or PBS was administered to the mice by IP injection once daily for 10 days.
In vitro myotube culture and treatments
The C2C12 mouse myoblast cell line was purchased from the American Type Culture Collection (Manassas, VA, USA) and grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% v/v fetal bovine serum (Invitrogen, Burlington, ON, Canada) and 1% penicillin/streptomycin at 37°C in 5% CO2. After growing to 100% confluence, cells were further cultured in DMEM containing 2% horse serum (differentiation medium [DM]) for 5 days to induce differentiation of myotubes. The first day of incubation in the DM was considered Day 0 of differentiation. The DM was changed every 24 hours. On Day 4 of differentiation, one of three DM-based media was added to wells containing the resulting C2C12 myotubes (in triplicate), and the plates were incubated for an additional 24 hours: (1) control group (0.02% v/v ethanol); (2) Dex group (10 μM Dex [D4902, Sigma-Aldrich]dissolved in ethanol); and (3) Dex + DLK1 group (10 μM Dex dissolved in ethanol with 5.0 μg/mL DLK1). After 24 hours growth, cells were harvested for extraction of total RNA and used for the analyses.
Tissue collection and histological analyses
Harvested TA tissue was fixed in 10%-PBS-buffered formaldehyde for 48 hours, embedded in paraffin, and sectioned into 5-μm-thick slices for Masson’s trichrome staining. After sealing the slides with neutral gum, the Masson’s trichrome-stained tissue slices were examined at 200× magnification. Blue-stained fibrosis area in Masson’s trichrome staining were quantified using the Image J software (NIH Image, Bethesda, MD, USA). After adjusting for the threshold within the section image, the surface area above the threshold was measured to determine the absolute and percentage area of blue-stained tissue.
Body composition analyses
On the day before the experiment was completed, four mice from each group were selected and subjected to dual energy X-ray absorptiometry (DXA, InAlyzer Medikors, Seongnam, Korea) for whole body composition analysis. The DXA scanner uses two separate low-dose X-ray exposures to read bone and soft tissue mass with a high degree of precision. Prior to the start of the experiment, the system was calibrated according to the manufacturer’s instructions. Software integrated into the scanner was used for data analysis.
Transmission electron microscopy
Two mice in each group with body weights similar to the median were selected as representative mice. A minimum of five tissue sites was randomly selected for imaging, and a representative image is presented in the figures. Samples of TA muscle were fixed with 2% glutaraldehyde in paraformaldehyde (Merck & Co. Inc., Kenilworth, NJ, USA) overnight at 4°C and post-fixed for 1 hour in 1% OsO4 resin (Polysciences Inc., Warrington, PA, USA). Samples were dehydrated in ethanol (Merck & Co.), embedded in rubber molds with epoxy resin (Polysciences Inc.), and polymerized in an oven at 60°C for 20 hours. Semithin sections (1 μm thick) were cut using a Leica Ultracut UCT (Leica Microsystems Inc., Buffalo Grove, IL USA) and stained with toluidine blue (Merck). Ultrathin sections (70 nm thick) were cut and mounted on coated copper grids (Nisshin EM, Tokyo, Japan) and double stained with 6% uranyl acetate (Ted Pella Inc., Redding, CA, USA) and lead citrate (Wako, Osaka, Japan). The ultrastructure of the tissue sections was observed using a JEM-1011 transmission electron microscope (TEM, JEOL, Tokyo, Japan) with an accelerating voltage of 80 Kv. Images were viewed using Camera-Megaview III software (EMSIS, Munster, Germany).
Quantitative real-time polymerase chain reaction
Total RNA was extracted using a Hybrid-R™ RNA purification kit (GeneAll Biotechnology, Seoul, Korea) according to the manufacturer’s instructions. Synthesis of cDNA from 1.0 μg total RNA per sample was performed in a 20 μL reaction volume using a high-capacity cDNA reverse transcription kit (Life Technologies, Carlsbad, CA, USA) and 2.5 μM of random primers. Quantitative real-time polymerase chain reaction (qPCR) was performed in 10 μL reaction volumes containing cDNA, 5 pmol of each oligonucleotide primer, and 5.0 μL of Power SYBR® Green PCR Master Mix (Life Technologies, Warrington, UK). The qPCR reactions were performed in a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) in a 96-well plate. The 2-ΔΔCt method was used to determine the relative abundance of mRNAs for muscle atrophy F-box protein (atrogin1), dynamin-related protein-1 (Drp1), myosin heavy chain (MHC) I, IIb, and IIx, MuRF1, myogenic differentiation (MyoD), myogenin, myostatin, and optic atrophy protein 1 (Opa1) using 18S ribosomal RNA (18S) as an internal reference. The mouse cDNA sequences were obtained from GeneBank (Supplemental Table S1).
Myostatin siRNA knock-down test
Double-stranded small interfering RNAs (siRNAs) targeting myostatin (20 nM) were purchased from QIAGEN (Cambridge, MA, USA). The C2C12 cells were transfected with siRNAs complexed with Lipofectamine™ 2000 reagent or negative control siRNA (Invitrogen, Carlsbad, CA, USA) according to manufacturer instructions. Myostatin siRNA transfection of myotubes was performed on Days 0 and 3 of differentiation. knockdown of myostatin gene expression (reduction ≥70%) was tested using the transfected cell line. After sufficient transfection, C2C12 cells were washed with PBS, the growth medium was changed on cell differentiation Day 4, and the wells were divided into three groups: (1) control group (0.02% v/v ethanol); (2) Dex group (10 μM Dex [D4902, Sigma-Aldrich] dissolved in ethanol); and (3) Dex+DLK1 group (10 μM Dex dissolved in ethanol with 5.0 μg/mL DLK1). After 24 hours, cells were harvested for Western blot analyses.
Western blot analyses
The C2C12 cells were grown for 4 days, with the DM changed every 24 hours. On the day of the experiment, fresh medium with test agents was added to the wells as follows. Cells were washed with PBS, scraped off in ice-cold PhosphoSafe™ buffer (Novagen, Merck), and centrifuged for 5 minutes at 16,000×g (4°C). Protein concentrations were determined from clear supernatants using a Bradford assay (Sigma-Aldrich) with bovine serum albumin as the control. Equal amounts (20 μg) of total protein were electrophoresed in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) 10% gradient gels. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). Samples were immunoblotted overnight with the indicated primary antibodies (typically at a 1:1,000 dilution) followed by secondary antibody conjugated with horseradish peroxidase (1:5,000 dilution). A SuperSignal™ West Pico PLUS kit (Thermo, Waltham, MA, USA) was used for detection. Proteins of interest were detected with the following specific antibodies: myogenin (F5D, Abcam, Boston, MA, USA), GDF8/myostatin (Abcam), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 6C5, Santa Cruz Biotechnology Inc., Dallas, TX, USA).
Statistical analyses
All results are presented as the mean±standard error of the mean. Statistical significance was calculated using a one-way analysis of variance (ANOVA) to assess differences between the multiple groups. Additionally, the Student’s t test was performed to compare values between two groups in post hoc analyses. A P<0.05 was considered statistically significant. All experiments were repeated at least three times. All statistical procedures were conducted using SPSS software version 25.0, for Windows (IBM Corp., Armonk, NY, USA).
RESULTS
Effects of DLK1 on body composition in the dexamethasone-induced muscle atrophy mouse model
The DXA body composition of representative mice from the three groups in the Dex-induced muscle atrophy model was analyzed (Fig. 1A–D). Body weight and bone volume did not differ between the three groups (Fig. 1A, B), but the percentage of tissue fat was significantly increased in the Dex group compared to the control group (P<0.05) (Fig. 1C). Lean body mass was significantly reduced in the Dex group compared to the control group, and DLK1 administration reversed the reduction in lean body mass which was evident in the Dex-only group (P<0.01) (Fig. 1D).
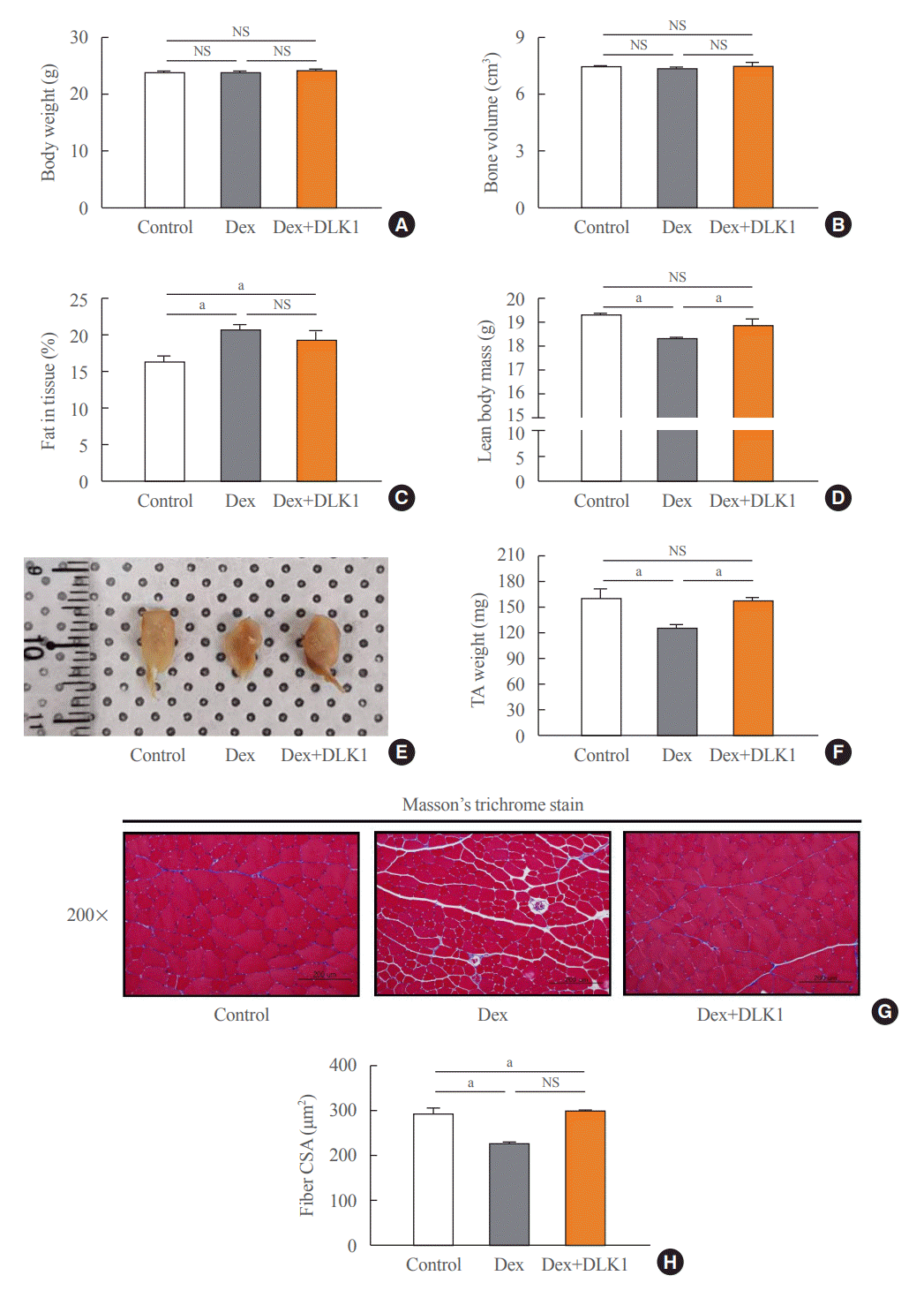
Fig. 1.
Effects of delta-like 1 homolog (DLK1) on body composition and histopathological morphology of tibialis anterior muscle in the dexamethasone-induced muscle atrophy mouse model. (A, B, C, D) Effects of DLK1 on body composition in dexamethasone-induced tibialis anterior (TA) skeletal muscle atrophy mice using dual energy X-ray absorptiometry (n=4 per each group). (E) Size comparison of TA muscles from a mouse hindlimb. (F) TA muscle weights (n=6 per each group). (G) Representative histopathological images of TA muscle (200× magnification; scale bar 200 μm) using Masson’s trichrome stain. (H) Mean cross-sectional area of the TA muscle (μm2). Results are presented as mean±standard error of the mean. Treatment groups were as follows: control=oral distilled water and intraperitoneal (IP) phosphate buffered saline (PBS); dexamethasone (Dex)=oral Dex (1 mg/kg) and IP PBS; Dex+DLK1=oral Dex (1 mg/kg) and IP DLK1 (0.8 mg/kg/day). CSA, cross-sectional area; NS, non-significant. aP<0.05 was considered statistically significant.
![]()
We also compared the TA muscle weight of mice from the three groups (Fig. 1E, F). The TA muscle weight was significantly lower in the Dex group than the control and Dex+DLK1 groups (P<0.05), but there was no difference between the control and Dex+DLK1 groups (Fig. 1F). Trichrome staining revealed reduced muscle fibril size and atrophy in the Dex group (Fig. 1G). Histology studies (Fig. 1H) indicated reduction of the cross-sectional area of TA myofibrils in the Dex group compared to the control group (22.3% reduction, P<0.05) (Fig. 1H), while the cross-sectional area of myofibrils in the Dex+DLK1 group was similar to that of the control group. Fibrotic area of TA muscle in the Dex group was significantly increased compared to the control group (Supplemental Fig. S1). DLK1 administration reduced Dex-induced fibrotic area, but statistical significance was not found. A significant reduction in mRNA expression was observed for type IIb fiber in the Dex group compared to the control group (Supplemental Fig. S2). In general, the relative mRNA expression ratio of each type of muscle fiber was consistent from 0.7 to 0.8 in the Dex group versus the control group, irrespective of fiber type. The relative expression of each type of fiber was not significantly different between the control and Dex+DLK1 groups.
Effects of DLK1 treatment on the expression of myostatin and muscle-related factors in dexamethasone- or cardiotoxin-induced muscle atrophy
In the TA muscle from the Dex-induced muscle atrophy model, mRNA expression of myostatin, a negative regulator of muscle mass that upregulates muscle-atrophy-related factors such as atrogin1 and MuRF1 [8], was up to three-fold greater than that of the control group, an effect that was significantly inhibited by DLK1 treatment (Fig. 2). Similarly, the expression of atrogin1 and MuRF1 was significantly greater in the Dex group compared to the control group, but these changes were not evident in the Dex+DLK1 treatment group (Fig. 2). In contrast, the expression of the myogenic factors MyoD and myogenin was significantly reduced in the Dex group, and these changes were attenuated in the Dex+DLK1 group (Fig. 2).
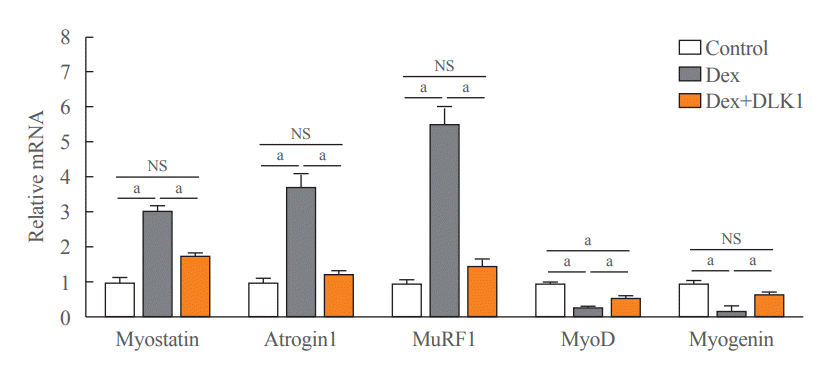
Fig. 2.
Effects of delta-like 1 homolog (DLK1) on muscle atrophic and myogenic factors in the dexamethasone-induced muscle atrophy mouse model. A graph showing relative mRNA expression of muscle atrophic (myostatin, atrogin1, and muscle-specific RING finger protein 1 [MuRF1]) and myogenic (myogenic differentiation [MyoD] and myogenin) factors in the mouse tibialis anterior (TA) muscle. Results are presented as mean±standard error of the mean. Treatment groups were as follows: control=oral distilled water and intraperitoneal (IP) phosphate buffered saline (PBS); dexamethasone (Dex)=oral Dex (1 mg/kg) and IP PBS; Dex+DLK1=oral Dex (1 mg/kg) and IP DLK1 (0.8 mg/kg/day). NS, non-significant. aP<0.05 was considered statistically significant.
![]()
In the CTX-induced atrophy model, TA muscle weight was tended to be lower in the Dex group than in the control and Dex+DLK1 groups on the tenth day after the CTX injection (Supplemental Fig. S3A). In the CTX model, the myostatin expression was greater in the Dex group than in the control group on the 10th day after the CTX injection, and DLK1 treatment significantly attenuated these changes (Supplemental Fig. S3B). The expression level of myogenin was higher in the Dex+DLK1 group than in the Dex group on the 5th day after the CTX injection (Supplemental Fig. S3C).
Effects of DLK1 on the morphology of myofibrils and associated organelles in dexamethasone-induced muscle atrophy
The effects of Dex and DLK1 administration on the inter-myofibrillar morphology and organelles (myofibril, Z line, and mitochondria) of the TA muscle were assessed using TEM (Fig. 3A). In contrast with the intact mitochondrial structure and cristae in the control group, swollen mitochondria with disorganized and fragmented cristae were evident in the Dex group. In the most severely damaged muscle cells, near complete dissolution of the internal architecture of both the Z line and myofibril was found. In the Dex+DLK1 group, Dex-induced mitochondrial swelling and disarray of the cristae were markedly reduced compared to the Dex-only group. The sarcomeres within TA muscle were significantly shorter in the Dex group than the control group (P<0.05) (Fig. 3B), while there was no difference in sarcomere length between the control and Dex+DLK1 groups. The mitochondria within the TA muscle were significantly longer in the Dex group than the control group (Fig. 3C) (P<0.05), and mitochondria lengthening was less pronounced in the Dex+DLK1 group. Regarding the mitochondria-shaping factors (Opa1 and Drp1) that regulate mitochondrial fusion and fission processes (Fig. 3D), decreased expression of Opa1 and increased expression of Drp1 were found in TA muscle from the Dex group compared to the control group. In contrast, in the Dex+DLK1 group, mitochondrial fusion-fission machinery in the TA muscle was relatively maintained.
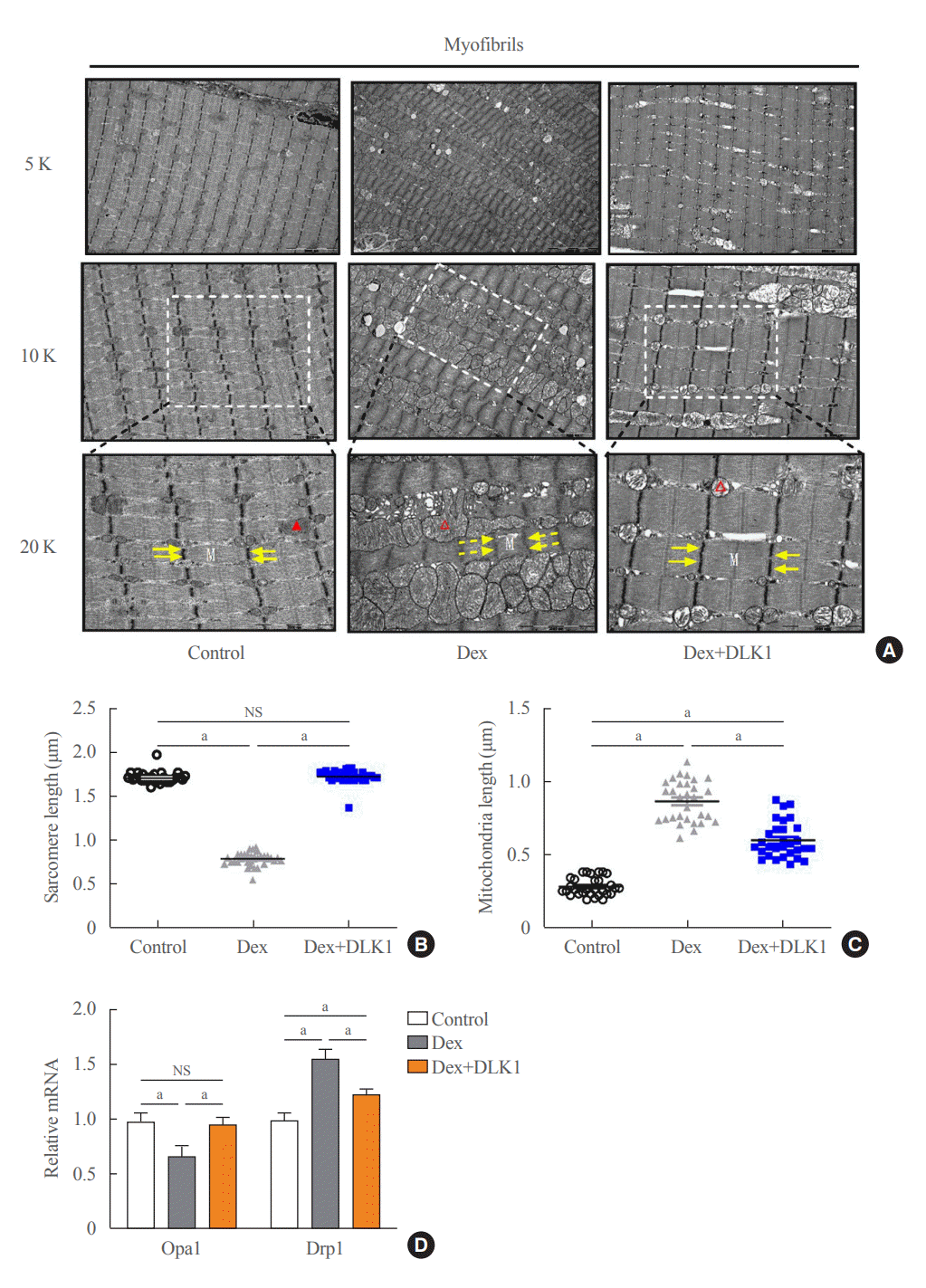
Fig. 3.
Effects of delta-like 1 homolog (DLK1) on tibialis anterior muscle morphology and mitochondrial biogenesis markers in the dexamethasone-induced muscle atrophy mouse model. (A) Representative electron microscopic images of myofibrils within the tibialis anterior (TA) muscle (5,000×, 10,000×, and 20,000× magnification; M=myofilament, yellow arrow=Z line, red arrow=mitochondria, dashed arrow=a broken structure, hollow red arrow=a swollen structure). Graphs showing lengths of (B) sarcomeres and (C) mitochondria in TA muscle cells (n=30 per each group). (D) Relative mRNA expression of mitochondrial fusion (Opa1) and fission (Drp1) markers (n=10 per each group). Results are presented as mean±standard error of the mean. Treatment groups were as follows: control=oral distilled water and intraperitoneal (IP) phosphate buffered saline (PBS); dexamethasone (Dex)=oral Dex (1 mg/kg) and IP PBS; Dex+DLK1=oral Dex (1 mg/kg) and IP DLK1 (0.8 mg/kg/day). NS, non-significant. aP<0.05 was considered statistically significant.
![]()
The TEM imaging of the TA muscle from the CTX-induced muscle atrophy model revealed no CTX-induced mitochondrial degeneration in the Dex+DLK1 group (Supplemental Fig. S3E).
Effects of DLK1 on dexamethasone-induced atrophy of myotubes
Dex was added into the culture medium of differentiated C2C12 myotubes for 24 hours on Day 4 of differentiation and resulted in myotube atrophy (Fig. 4A). In contrast, myotube thickness was significantly increased by the addition of DLK1 (5 μg/mL) to Dex-treated C2C12 myotubes compared to the addition of Dex alone (Fig. 4A, B). The C2C12 myotubes incubated with Dex showed a significant increase in the mRNA expression of myostatin and of muscle atrophy factors, atrogin1 and MuRF1, compared to ethanol-treated controls (Fig. 4C). In addition, Dex suppressed mRNA expression of the myogenic factors, MyoD and myogenin. In contrast, the addition of DLK1 to Dex prevented the atrophic effect of Dex in C2C12 myotube cultures by reducing the expression of muscle atrophy factors and increasing the expression of myogenic factors. Incubation with Dex also induced structural and morphological changes in myotubular mitochondria, characterized by elongation and swelling of mitochondria, but these changes were not evident in cultures to which Dex and DLK1 were added (Fig. 5A). Along with mitochondrial morphological changes, Dex significantly reduced mRNA expression of the fusion regulator Opa1 and increased mRNA expression of the fission regulator Drp1 compared to those of the control (Fig. 5B). Addition of Dex+DLK1 to myotube cultures normalized Dex-induced changes in the expression of mitochondrial fusion and fission markers to levels comparable with those of the control group (Fig. 5B). To confirm whether the protective effect of DLK1 on muscle atrophy is mediated by modulation of myostatin, myostatin expression was down-regulated in differentiated C2Cl2 myotubes using siRNA transfection. In myotubes transfected with negative control siRNAs, Dex treatment significantly increased myostatin expression and decreased myogenin expression (Fig. 6); these effects were not evident in the Dex+DLK1 myotube cultures, which showed comparable myostatin and myogenin expression to the control group. However, under the condition of myostatin knockdown, no significant difference in myostatin and myogenin protein expression were found in the Dex-treated myotube cultures compared to the control and Dex+DLK1 myotube cultures (Fig. 6).
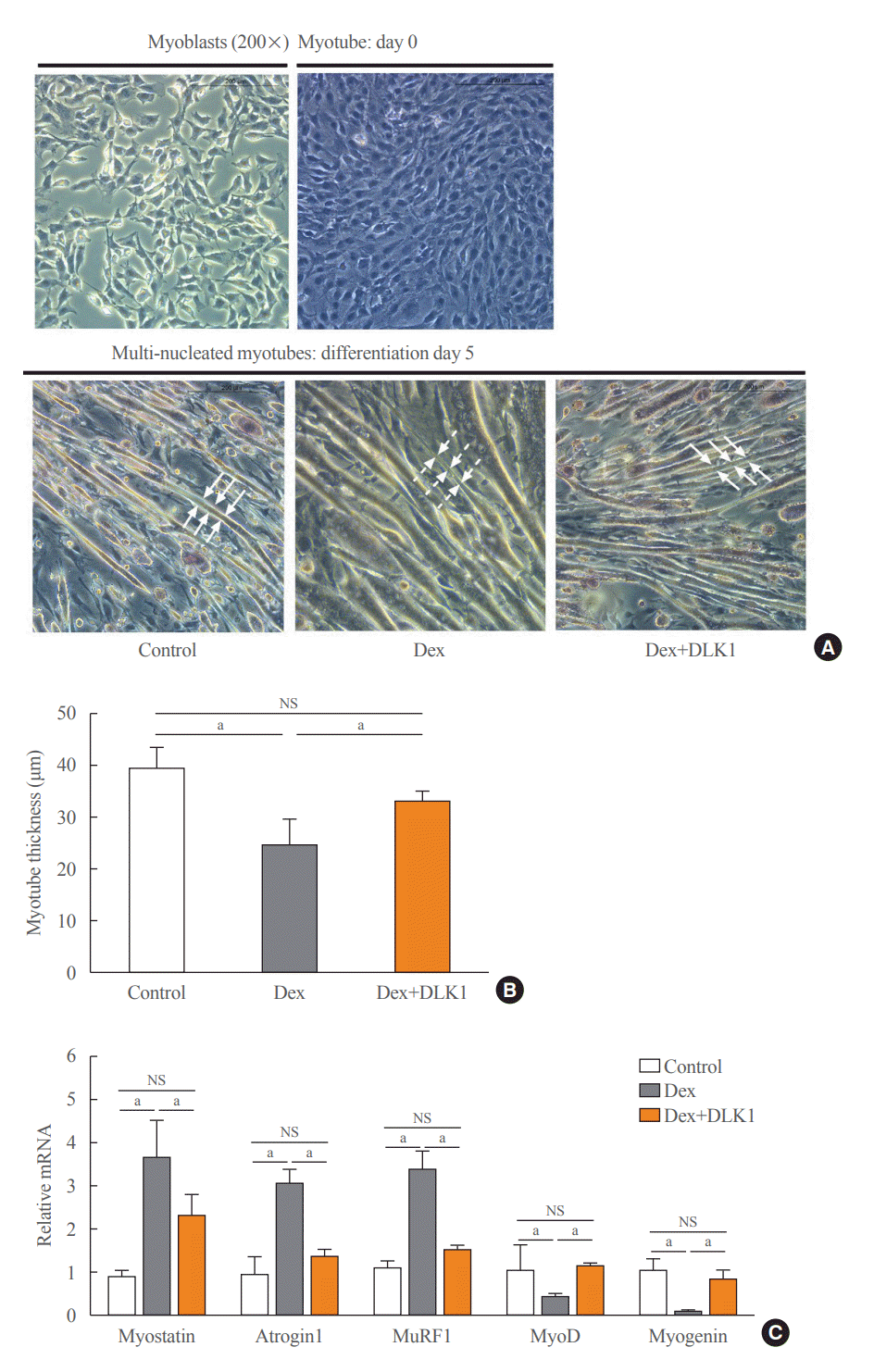

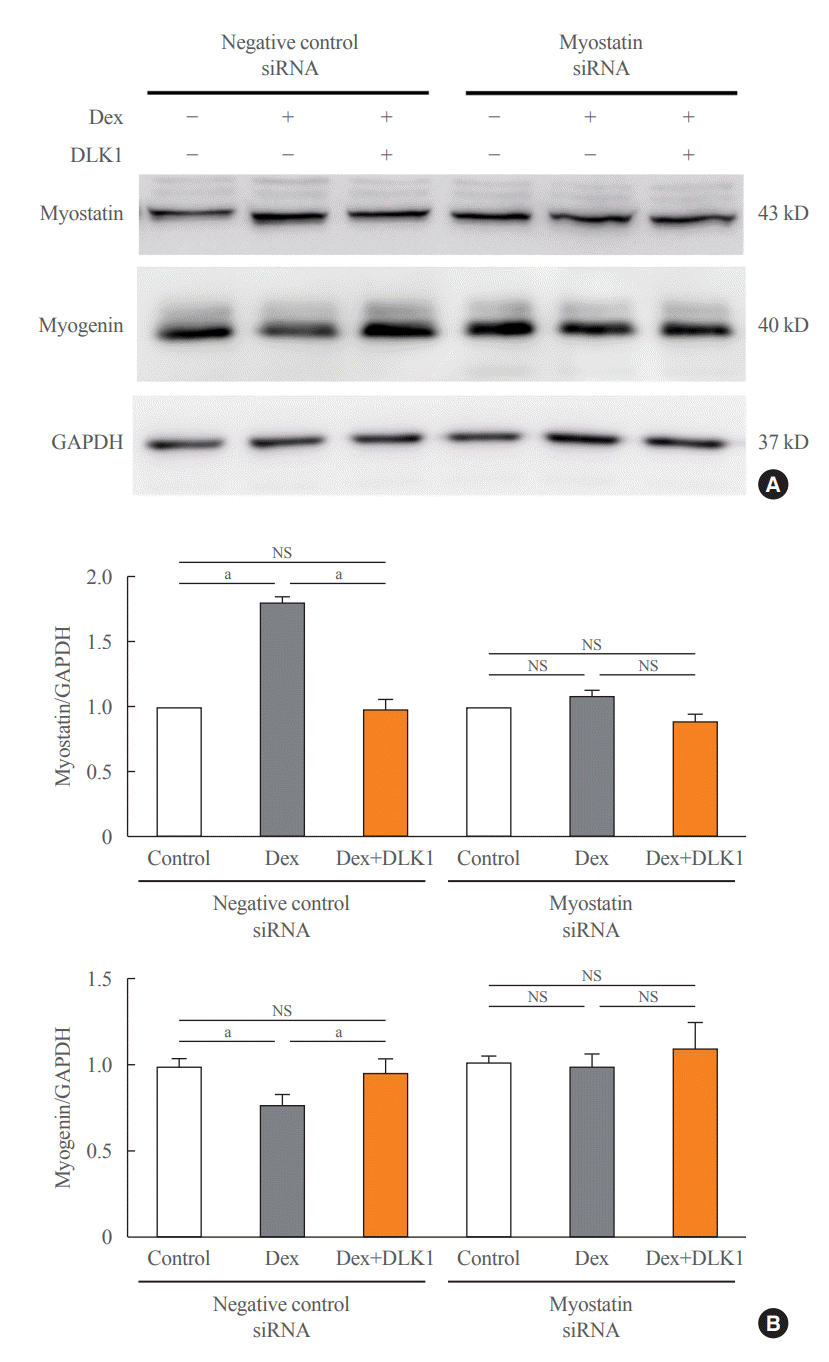
Fig. 4.
Effect of delta-like 1 homolog (DLK1) on dexamethasone-induced myotube atrophy. (A) Representative histopathological images of H&E staining showing myoblast (Day 0) and differentiated multi-nucleated myotubes (Day 5) according to the type of differentiation medium. (B) Thickness of myotubes (µm). (C) Relative mRNA expression of muscle atrophic (myostatin, atrogin1, and muscle-specific RING finger protein 1 [MuRF1]) and myogenic (myogenic differentiation [MyoD] and myogenin) factors in myotubes. Results are presented as mean±standard error of the mean. Myotube cultures were divided into three groups on differentiation Day 4 and cultured in different media for 24 hours: control=Dulbecco’s Modified Eagle Medium (DMEM)+ethanol (0.02% v/v); dexamethasone (Dex)=DMEM+Dex (10 μM dissolved in ethanol); and Dex+DLK1=DMEM+Dex (10 μM dissolved in ethanol)+DLK1 (5.0 μg/mL). NS, non-significant. aP<0.05 was considered statistically significant.
![]()
Fig. 5.
Effects of delta-like 1 homolog (DLK1) on mitochondrial biogenesis markers in dexamethasone-induced myotube atrophy. (A) Representative electron microscopic images of myotube cells (5,000×, 15,000×, and 40,000× magnification). (B) Relative mRNA expression of mitochondrial fusion (Opa1) and fission (Drp1) markers (n=10 per each group). Results are presented as mean±standard error of the
mean. Myotube cultures were divided into three groups on differentiation Day 4 and cultured in different media for 24 hours: control=Dulbecco’s Modified Eagle Medium (DMEM)+ethanol (0.02% v/v); dexamethasone (Dex)=DMEM+Dex (10 μM dissolved in ethanol); and Dex+DLK1=DMEM+Dex (10 μM dissolved in ethanol)+DLK1 (5.0 μg/mL). NS, non-significant. aP<0.05 was considered statistically significant.
![]()
Fig. 6.
Effect of delta-like 1 homolog (DLK1) on muscle-related markers in atrophied myotubes with or without knockdown of myostatin expression. (A) Western blot images with (B) quantification of myostatin and myogenin protein after transfection with small interfering ribonucleic acids (siRNAs) of negative control or myostatin (n=3 to 6 per group). Results are presented as mean±standard error of the mean. Transfected myotube cultures were divided into three groups on differentiation Day 4 and cultured in different media for 24 hours: control=Dulbecco’s Modified Eagle Medium (DMEM)+ethanol (0.02% v/v); dexamethasone (Dex)=DMEM+Dex (10 μM dissolved in ethanol); and Dex+DLK1=DMEM+Dex (10 μM dissolved in ethanol)+DLK1 (5.0 μg/mL). GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NS, non-significant. aP<0.05 was considered statistically significant.
![]()
DISCUSSION
In the present study, we demonstrated that DLK1 attenuated Dex- and CTX-induced muscular degenerative changes by decreasing myostatin expression in these mouse muscle atrophy models. In addition, DLK1 treatment improved mitochondrial dynamics in the Dex-induced atrophy model. Knockdown of myostatin reduced the protective effect of DLK1 against muscle atrophy in vitro, suggesting critical role for myostatin in DLK1-mediated muscle protection.
The balance between muscle proteolysis and synthesis is regulated by myokines, the cytokines secreted by muscle [10]. Myostatin is an important myokine that negatively regulates muscle hypertrophy [11]. Myostatin is a member of the transforming growth factor β (TGF-β) family and essential for negative regulation of skeletal muscle growth [12]. Mutation of myostatin was reported to lead to an increase in muscle mass in vivo [13]. Therefore, myostatin might be an attractive target for treatment of sarcopenia associated with muscle atrophy [14].
Attenuation of muscular atrophy process by DLK1 treatment in this study could be mediated by inhibition of myostatin downstream signaling, as DLK1 is a pharmaceutical composition for suppressing linkage between activin receptor type 2 B (ACVR2B; a TGF-β type II receptor) and its ligands, representatively myostatin [15,16] in a competitive manner (Supplemental Fig. S4). Binding of myosatin to ACVR2B stimulates the phosphorylation of Smad2/3 [15-17]. Phosphorylated Smad2/3 leads to down-regulation of myogenic genes such as MyoD and myogenin [18]. In addition to Smad2/3 stimulation, myostatin pathway inhibits Akt, which consequently activates forkhead box O (FoxO) signaling [15]. Activated FoxO upregulates transcription of atrophy-specific genes, such as atrogin1 and MuRF1 [15]. Furthermore, independent of regulating Smad, FoxO and Akt, myostatin signaling can interact with peroxisome proliferator-activated receptor-γ coactivator-1 α (PGC1-α) in muscle [15]. Suppression of myostatin is associated with increased activity of PGC1-α [15,19]. PGC1-α enhances mitochondrial biogenesis in muscle by upregulating mitochondrial fusion genes including Opa1 [19,20]. In oxidative stress condition, PGC1-α protects muscle cells from ROS-mediated mitochondrial removal [21].
Dex upregulates muscle degradation, suppresses muscle synthesis, and causes muscular atrophy [22]. Dex-induced muscle proteolysis is induced by stimulation of the ubiquitin proteasome system that is composed of muscle-specific proteins (the so-called atrogenes), including atrogin1 and MuRF1 [23]. Both MyoD and myogenin are myogenic regulatory factors [24]: MyoD upregulates skeletal myogenesis that directs contractile protein synthesis and mediates muscle injury repair process [25], and myogenin enhances muscular biosynthetic pathway and regulates differentiation of myoblasts to multinucleated myotubes under atrophic conditions [26]. Loss of the myogenin gene is a lethal mutation due to severe skeletal muscle deficiency as myoblasts are unable to fuse into multinucleated myofibers without myogenin activity [27]. Dex treatment is associated with reduced expressions of MyoD and myogenin in muscle, similar to catabolic states. In the present study, DLK1 administration attenuated Dex-induced muscle atrophy and loss of TA muscle weight. Consistent to previous findings [28], in this study, Dex upregulated expression levels of myostatin and muscular-atrophy-related factors including atrogin1 and MuRF1. These Dex-induced changes were attenuated by DLK1 treatment. In addition, Dex-induced reductions in MyoD and myogenin were inhibited by DLK1 treatment. In the present in vitro study using knockdown of endogenous myostatin expression [29], the protective effect of DLK1 against muscle atrophy was diminished, suggesting that DLK1 treatment inhibits atrophic progression through regulation of the myostatin signaling pathway [30].
The CTX-induced model of muscle injury involves disruption of ion fluxes, mediated by membrane depolarization, and is accompanied by the loss of protein content and organellar breakdown [31]. Myogenic regulatory factors including myogenin are sharply induced at Days 3 to 5 after CTX injury to promote MyoD in the regeneration process after injury [32]. In the present study, myogenin expression in the TA muscle was significantly amplified by DLK1 treatment on Day 5 after CTX injection, suggesting an augmented regenerative response to injury. Thus, we postulate that DLK1 generally enhances muscular regeneration after various injurious stimuli such as Dex and CTX. Further studies are needed to assess potential clinical applications of DLK1.
Muscular atrophy is characterized histologically by decreased muscle size and protein content, and loss of cellular organelles [33], accompanied by changes in muscle fibers and mitochondria. The mitochondrial architecture and function in skeletal muscle are maintained by fusion/fission machinery [34,35], and homeostatic regulation of mitochondrial fusion and fission by two transcription factors, Opa1 and Drp1, is disrupted in muscle wasting in vivo [35,36]. Fusion of the inner mitochondrial membrane is mediated by Opa1 [37], and loss of Opa1 is associated with age-dependent muscle loss and weakness [35]. In the present study, DLK1 treatment blocked Dex-mediated reductions of Opa1 in vitro and in vivo. Mitochondrial fission is an early event during cellular degeneration of muscle [37-39]. The mitochondrial fission factor Drp1 was increased by Dex, and DLK1 attenuated Dex-mediated Drp1 upregulation in vitro and in vivo. These results suggest that DLK1 treatment attenuated Dex-mediated muscle atrophy in our model by modulation of Opa1 and Drp1, preventing Dex-mediated alterations in mitochondrial fusion and fission homeostasis.
The balance between synthesis and degradation of myofibrillar protein determines muscle mass. When Dex and DLK1 were co-administered, the diameter of myotubes was significantly increased compared to the effects of Dex treatment alone. These changes were accompanied by ultrastructural changes in muscle fibers as assessed using TEM. In the TA muscle of mice with Dex-induced muscular atrophy, muscle cell mitochondria displayed swollen features, similar to the findings of a previous report [40], while DLK1 treatment partly blocked these changes in mitochondrial morphology and density. Based on these data, we postulate that changes in molecular markers of muscle atrophy, differentiation, and mitochondria induced by DLK1 treatment consequently lead to improvement in muscle architecture.
The current study has some limitations. First, aging-related biological changes mainly drive sarcopenia in humans [41], but DLK1 attenuated muscle wasting by Dex or CTX in the current study. Thus, further studies are needed to investigate the beneficial effect of DLK1 on aging-driven muscle atrophy in vivo. Second, the expression of myogenic factors including MyoD and myogenin was not measured daily after Dex or CTX injection. As muscle regeneration after CTX injury involves time-dependent phases of tissue healing [31], daily evaluation of myogenic factors could provide more detailed information about DLK1-mediated enhancement of muscle regeneration. Third, changes in gross and microscopic feature and muscular biomarkers were investigated using TA muscle, as we considered that muscular atrophy induced by Dex or CTX would be more prominent in the glycolytic muscles such as TA muscle, due to their more vulnerability to stressors such as aging and corticosteroid treatment than oxidative muscle such as soleus muscle [42,43]. However, the absence of data in the oxidative muscle limits the interpretation of DLK1’s effect on muscular atrophy. The effect of DLK1 on muscular atrophy needs to be further comprehensively validated using both glycolytic and oxidative muscles. Fourth, a positive correlation between DLK1 and insulin resistance was observed previously [44], but changes in insulin resistance-related parameters were not measured in this study. To safely use DLK1 in sarcopenia which is frequently accompanied with insulin resistance and diabetes [45], the effect of DLK1 on insulin resistance requires further investigation. Fifth, the capacity of DLK1 could be decided using the degree of increase in TA muscle and lean body mass in the present study. However, changes in muscle strength by DLK1 treatment should be additionally assessed using methods such as forelimb grip strength [46], as sarcopenia, a disease that DLK1 aims to treat, is defined as a progressive decline of not only skeletal muscle mass, but also muscle strength and functions [47,48]. Sixth, changes in mitochondrial function by DLK1 treatment was not investigated. As intact mitochondrial function is an important determinant to prevent muscle atrophy [49], further experiments such as high-resolution respirometry to measure mitochondrial function [50,51], are needed to support the benefit of DLK1 on sarcopenia.
In summary, the present study demonstrated that DLK1 administration has a therapeutic effect on muscular atrophy in vivo, and the action of DLK1 might be mediated by suppression of the myostatin-driven signal transduction system. Collectively, DLK1 could be a promising candidate for treatment of sarcopenia characterized by muscle atrophy and dysfunction.
 XML Download
XML Download