

 Citation
Citation Print
Print



INTRODUCTION
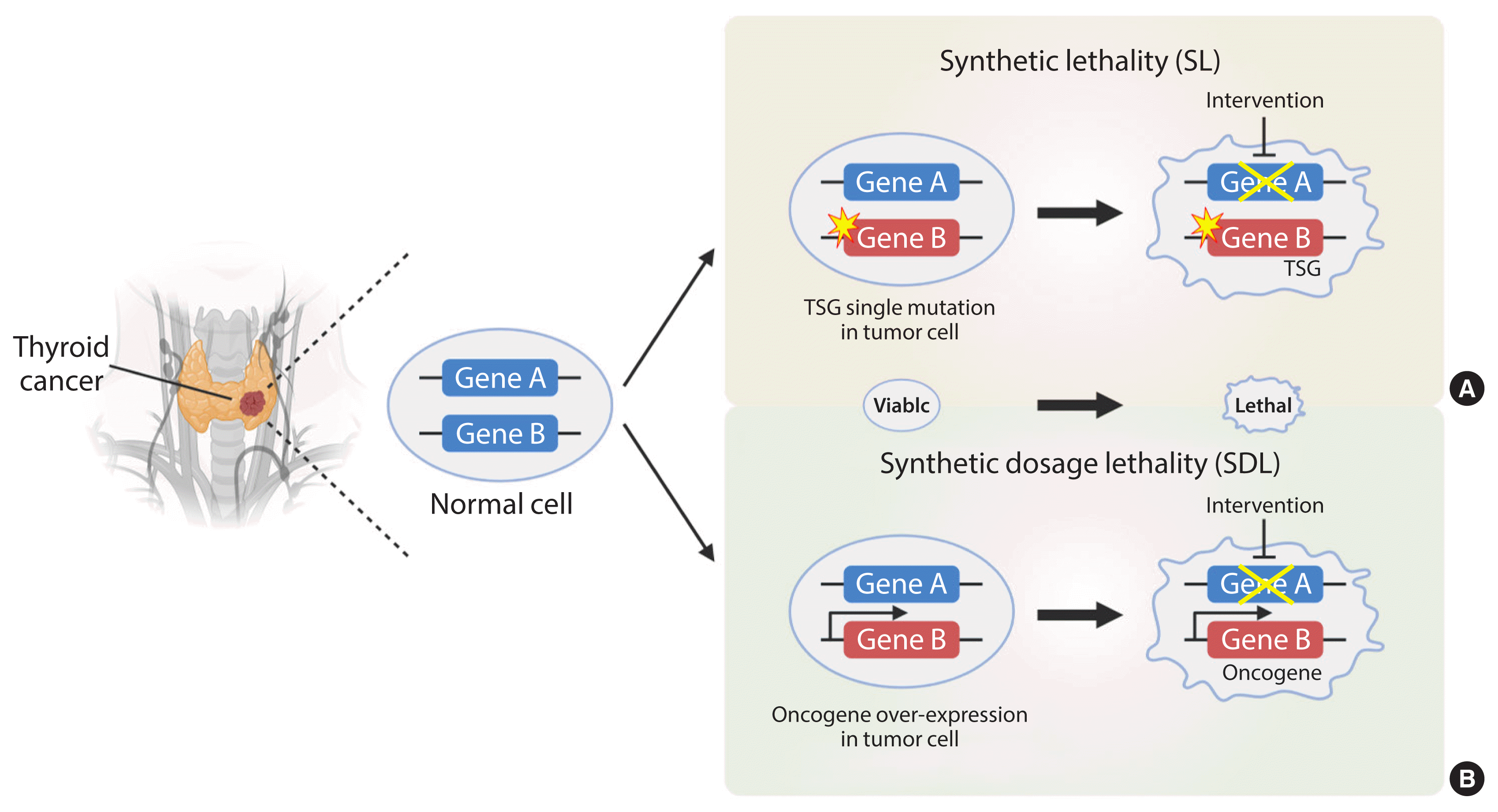
Synthetic lethality (SL), first proposed in 1922 by the American geneticist Calvin Bridges, is observed when simultaneous inhibition of two genes results in cellular lethality [1]. The meaning of synthetic used here conforms to the ancient Greek meaning: “the combination of entities to form something new.” Classical SL occurs when inhibition of two genes is lethal, while inhibition of either gene alone is not (Fig. 1). It can be harnessed to treat cancer selectively by identifying inactive genes and targeting their SL partner(s) [2]. In accordance with this concept, SL can also be applied to genes and small molecules used to elucidate the mode of action of drugs [3–5]. Cancer is driven by consequences of mutation or amplification of genes that allow cells to escape mechanisms that control proliferation and survival. Hence, inhibitors that target SL partners of mutated or amplified genes in cancer cells can cause cancer cell death without affecting the survival of normal cells.
The SL concept is divided into two categories due to its complexity: SL and synthetic dosage lethality (SDL); the latter is a slight deviation from the original concept of SL (Fig. 1). SL occurs in cancers caused by loss-of-function of tumor suppressors and their partner genes. This is a genetic interaction in which combination of two mutations leads to cell death, whereas a single mutation in either of the genes does not. SDL occurs during interaction between an oncogene and its interactor gene [6]. Principally, SL and SDL offer a unique opportunity to develop selective anticancer drugs [7,8].
Recent studies identified SL interactions via systematic screening of human cancer cells using small molecule inhibitors and high-throughput short hairpin RNA (shRNA) or Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-based screens [9,10]. These screens have been used successfully to identify SL pathways associated with known tumor suppressors and oncogenes [11]. Therapeutics developed using the SL approach have been used to treat breast cancer. Loss-of-function of breast cancer susceptibility genes 1 and 2 (BRCA1 and BRCA2), coupled with inhibition of poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP), is an SL interaction that results in marked sensitization of cancer cells [12,13]. Fusion oncogenes such as RET/papillary thyroid cancer (PTC) and breakpoint cluster region-Abelson (BCR-ABL) drive carcinogenesis in thyroid cancer and chronic myelogenous leukemia (CML). Addiction of tumor cells to constitutively active RET/PTC and BCR-ABL protein kinases results in sensitization to kinase inhibitors such as imatinib [14–16]. Recently, screening identified an SL interaction between signal transducer and activator of transcription 3 (STAT3) and BCR-ABL, such that combined inhibition of STAT3 and BCR-ABL1 induced SL in therapy-resistant CML [17].
Furthermore, targeting undruggable oncogenes such as KRAS mutations, which are common in refractory thyroid cancer, can be approached via SL interactions [18–20]. MYC oncogenes are implicated in the development of a large number of cancers, including thyroid cancer [21]. Although MYC is an attractive target for cancer therapeutics, lack of defined pockets in the MYC protein mean that it is regarded traditionally as undruggable by low molecular weight inhibitors [22]. A recent study aimed at identifying SL partners of oncogenic MYC revealed that the most over-represented functional categories among MYC SL genes are DNA-repair and the cell cycle [23–26]. Elucidation of KRAS and MYC synthetic lethal interactions is still in its infancy, and how these interactions are influenced by tissue-specific programs and by concurrent genetic changes requires further investigation. Nevertheless, we predict that these studies may lead to novel therapeutic approaches and new insights into the role of KRAS and MYC in thyroid cancers that are refractory on radioiodine and kinase inhibitors.
METABOLIC SYNTHETIC LETHALITY
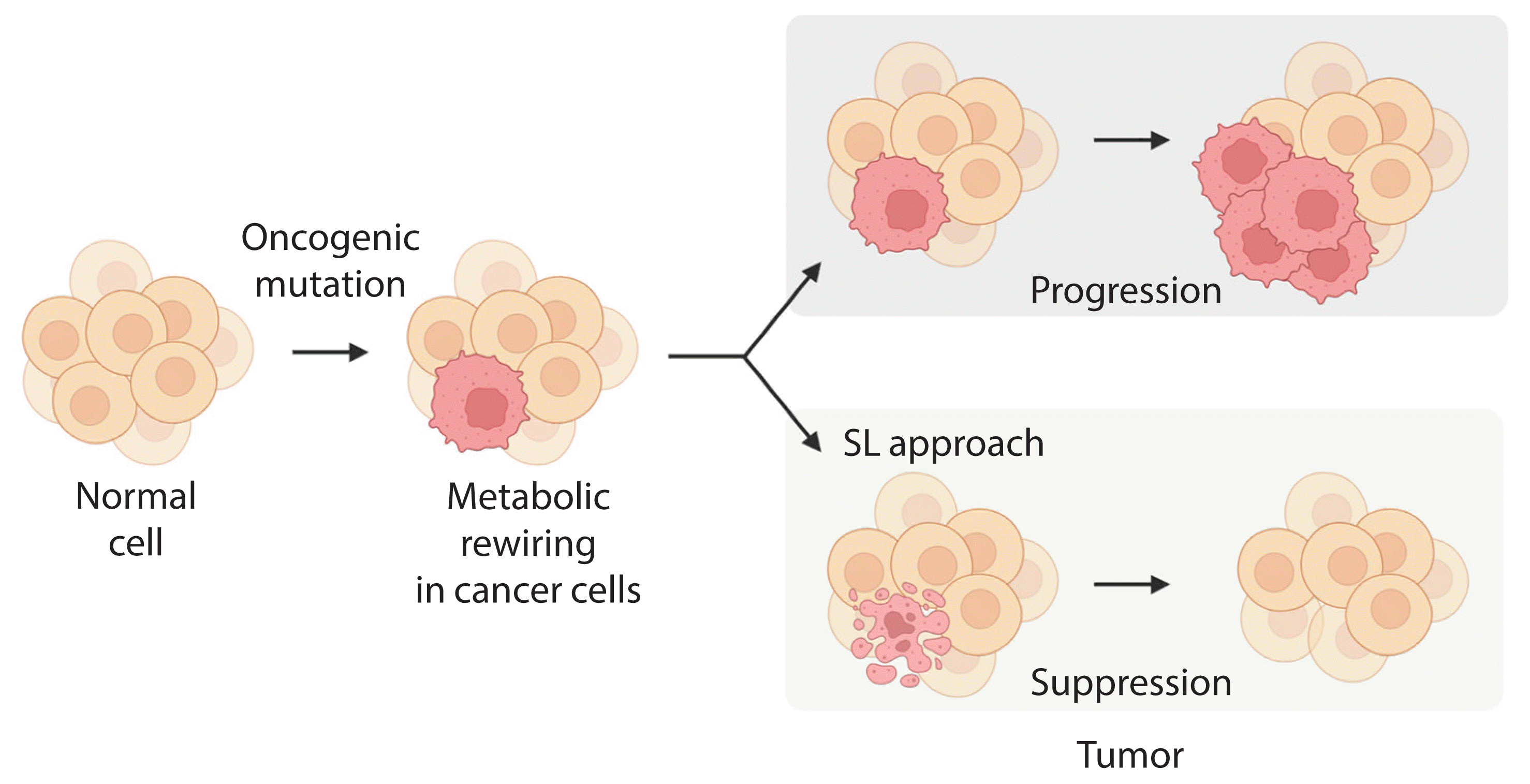
Altered cellular metabolism is a hallmark of cancer, and a deeper understanding of cancer cell metabolism may provide the basis for metabolism-based cancer therapy. This concept was encouraged by the finding that oncogene mutations, or mutations in key metabolic enzymes, drive tumorigenesis by reprogramming or rewiring cell metabolism required for dysregulated cell growth and proliferation [27]. Cancer cells largely use three key metabolic processes, glycolysis, amino acid catabolism, and mitochondrial oxidative phosphorylation (OXPHOS), to adapt to the increased energy demand required to drive growth and metastasis [27,28]. Recently, the concept of SL was expanded to include cellular metabolism, and it was applied to cancer-specific metabolic remodeling (Fig. 2) [29–32]. For example, AMPK-related kinase 5 (ARK5), an upstream regulator of AMP-activated protein kinase (AMPK), is an SL interactor with oncogenic MYC via activation of the mammalian target of rapamycin (mTOR) pathway [25]. In oncogenic MYC-expressing cells, inhibition of ARK5 leads to a collapse in adenosine triphosphate (ATP) levels and results in induction of pro-apoptotic responses. In addition, depletion of ARK5 in MYC-driven mouse models of hepatocellular carcinoma prolongs survival and demonstrates the therapeutic value of this SL interaction.
Enhanced glucose uptake and an increased rate of glycolysis are major remodeled metabolic features of cancer cells, including differentiated and undifferentiated thyroid cancers. Increased flux of glucose into cancer cells generates high levels of lactate from pyruvate through lactate dehydrogenase A (LDHA) [33,34]. Blockade of LDHA is not sufficient to inhibit cancer cells, but it remodels amino acid biosynthesis by engaging activating transcription factor 4 (ATF4)-dependent metabolic reprogramming and by inducing solute carrier family 1 member 5 (SLC1A5) to increase uptake of glutamine and essential amino acids, thereby enhancing mammalian target of rapamycin complex 1 (mTORC1) activity and cell proliferation. Co-targeting LDHA and mTORC1 has a synergistic effect with respect to suppressing melanoma cell proliferation and tumor growth [35]. In addition, activation of the LDHA pathway reduces nicotinamide adenine dinucleotide (NAD+) to NADH, which is used to transport electrons to the electron transport chain (ETC). The combination of LDHA inhibition with small inhibitors FX11 and FK866 (NAD+ synthesis inhibitors) reduces the NAD+ cellular pool in lymphoma, resulting in tumor regression [36]. Dual inhibition of glycolysis and mitochondrial function using a combination of 2-deoxyglucose, a specific glycolysis inhibitor, and metformin impairs tumor growth in mouse xenograft models [37,38]. Recently, Benjamin et al. [39,40] showed that syrosingopine, a dual monocarboxylate transporter 1 (MCT1) and MCT4 inhibitor that prevents lactate and H+ efflux, elicits SL in combination with metformin. This finding indicates that loss of NAD+ regeneration capacity due to combined treatment with metformin and syrosingopine results in glycolytic blockade, leading to ATP depletion and cell death. A previous study shows that metformin has an in vitro synergistic anticancer effect against anaplastic thyroid cancer when used in combination with vemurafenib [41].
The serine synthesis pathway and one-carbon metabolism are often dysregulated in cancer, including thyroid cancer, due to their contribution to cellular building blocks, DNA methylation, post-translational modification, and redox homeostasis [42]. The complex networks of enzymes such as phosphoglycerate dehydrogenase (PHGDH), serine hydromethyl transferase (SHMT), and methylenetetrahydrofolate dehydrogenase (MTHFD), which are required for the serine synthesis pathway and one-carbon metabolism in the cytoplasm and mitochondria, may provide a target for SL in thyroid cancer [43].
SL screens targeting metabolic enzymes could be useful tools that might add to the number of metabolic targets for anticancer therapy, and they could be exploited to overcome resistance to conventional chemotherapy agents such as bevacizumab (Avastin, Genentech Inc., San Francisco, CA, USA). A potential target is glycogen phosphorylase liver form (PYGL), expression of which increased following bevacizumab treatment in an in vivo xenograft model [44]. Combination treatment with inhibitors targeting PYGL and/or other hypoxia-inducible factor (HIF)-dependent metabolic enzymes essential for survival under hypoxic conditions, plus an anti-angiogenic drug, could be of great interest as a metabolic cancer therapy.
Alteration of one single pathway in the metabolic network may induce compensatory pathways to generate alternative sources that compensate for the loss of certain metabolites. Metabolic SL occurs when simultaneous mutations in two different metabolic genes are lethal, while a mutation in either of the individual genes alone is tolerable. The simultaneous suppression of several genes that together have the potential for SL may open new avenues for anticancer treatment. The genome-scale network model of cancer metabolism, shRNA, and CRISPR/Cas9 screening to validate SL, will be used widely to identify genes essential for cancer cell proliferation. Furthermore, they will predict combinations of SL drug targets that can be used to treat thyroid cancer.
Understanding the context-dependent functions of SL allows therapeutic targeting of cancer. Furthermore, elucidating SL gene combinations could identify clinically relevant drug combinations, as well as biomarkers, that will allow development of improved treatments. The recent SL gene combinations used in preclinical and clinical settings may facilitate development of new therapeutic approaches and identify potential biomarkers [45].
BRAF MUTATION AND SDL
The BRAFV600E mutation is the most common genetic alteration in thyroid cancer, occurring in about 45% of sporadic PTCs, particularly relatively aggressive subtypes such as the tall-cell PTC [46]. The BRAFV600E mutant protein has 500-fold greater kinase activity than the wild-type [47], and this mutation is mutually exclusive with other common genetic alterations, supporting its independent oncogenic role (as demonstrated by targeted expression studies showing BRAF mutation-initiated development of PTC and its transition to anaplastic thyroid cancer) [48,49].
Several tyrosine kinase inhibitors have been approved for treatment of differentiated and anaplastic thyroid cancers. Lenvatinib and sorafenib are used widely to treat radioiodine-refractory differentiated thyroid cancer. However, although these targeted therapies extend median progression-free survival, they do not increase overall survival [50]. A significant number of severe side effects, as well as development of drug resistance, prevent effective and long-term treatment. Additional studies are needed to characterize the molecular and metabolic landscape, and to identify a potential effective therapy for aggressive BRAFV600E mutant thyroid cancers.
The high prevalence of the BRAF mutation makes it an attractive SDL lethal target due to possible signaling dependencies consequent to hyperactivation of the mitogen-activated protein kinase (MAPK) pathway. Transcription factor networks downstream of MAPK, as well as cataloged MAPK target genes encoding nuclear DNA-binding proteins, have been identified in colorectal cancer cell lines [51,52]. Klotz-Noack et al. [53] performed a shRNA screen to probe MAPK targets encoding nuclear and/or DNA-binding factors in isogenic colorectal cell lines inducible for oncogenic BRAF. They identified the multifunctional Splicing Factor Proline and Glutamine Rich Protein (SFPQ) as a novel factor that is SL with BRAFV600E. Depletion of SFPQ from BRAFV600E-expressing, but not isogenic BRAF-wild-type, colorectal cancer cells halted cell cycle progression at S phase and triggered apoptotic cell death [53]. Mechanistically, loss of SFPQ from BRAF-mutant cancer cells triggers the Chk1-dependent replication checkpoint, resulting in decreased cell numbers and reduced activity of replication factories, thereby increasing collision between replication and transcription [53]. BRAFV600E-mutant cancer cells and organoids are sensitive to combinations of Chk1 inhibitors and chemically-induced replication stress, pointing toward future therapeutic approaches that exploit nuclear vulnerabilities induced by BRAFV600E [53]. This screening approach highlights an underused approach to targeted therapeutic interference in BRAF-mutant colorectal cancer cells by exploiting the essential nuclear programs induced by MAPK activity identified in thyroid cancer cell lines. Consistent with the results in colon cancer cell lines, low expression of SFPQ is associated with decreased survival probability in The Cancer Genome Atlas (TCGA) dataset [54]. Further investigation of the role of SFPQ in the context of BRAFV600E-mutant thyroid cancer cells is required. Publicly available cancer cell lines and tumor tissue genomic data used to look for SDL partners of mutant BRAF identified limited SL pairs [55]. The genes PCDHGB4, ZNF138, CXCR2, CDH2, and DGKA were suggested as potential SL genes in BRAF-mutant cancer cells [55].
Li et al. [56] identified cytochrome P450 family 2 subfamily S member 1 (CYP2S1) as a SL partner of BRAFV600E in thyroid cancer. They found that expression of CYP2S1 in PTCs, particularly in conventional and tall-cell PTCs, is higher than that in normal thyroid tissues, and that its expression is positively associated with the BRAFV600E mutation. Consistent with this, knockdown of CYP2S1 selectively inhibits cell proliferation, migration, invasion, and tumorigenic potential in nude mice, and promotes cell apoptosis in BRAFV600E-mutated thyroid cancer cells, but not in BRAF wild-type lines. Mechanistically, the BRAFV600E-mediated MAPK/extracellular signal-regulated kinase (ERK) cascade upregulates CYP2S1 expression via an aryl hydrocarbon receptor (AHR)-dependent pathway, while CYP2S1 in turn enhances the transcriptional activity of AHR through its metabolites [56].
The metabolic remodeling induced by BRAFV600E expression can also be exploited as a SL vulnerability [57]. Use of an shRNA screen selectively targeting genes involved in ketogenic metabolism in BRAFV600E and BRAF wild-type melanoma cell lines identified 3-hydroxy-3-methylglutaryl-CoA lyase (HMGCL) as a possible SL partner for BRAFV600E [57]. Increased levels of HMGCL, an enzyme involved in ketogenesis, were observed in BRAFV600E melanomas in which HMGCL was knocked down, resulting in decreased growth solely in BRAFV600E mutant lines. Mechanistically, acetoacetate (the metabolite created by HMGCL) increases binding of BRAFV600E to MAPK kinase (MEK), with subsequent phosphorylation and activation of the MAPK pathway [57]. Suppression of HMGCL specifically attenuates proliferation and growth potential caused by decreased MAPK pathway activity. Data from the human protein atlas dataset show that thyroid cancer has weak to moderate cytoplasmic immunoreactivity for HMGCL. Therefore, the susceptibility of BRAFV600E thyroid cancer cells to inhibition of HMGCL remains to be determined.
BRAF-induced senescence is a tumor suppressive mechanism that prevents transformation in response to constitutive oncogene activation. Activation of oncogenes also promotes metabolic reprogramming to support cancer cell metabolism, but the role of metabolic rewiring in BRAFV600E-induced senescence remains to be identified. Kaplon et al. [58] showed that cells undergoing BRAFV600E-induced senescence exhibited increased incorporation of pyruvate into the tricarboxylic acid (TCA) cycle, as well as increased redox stress. This increase in mitochondrial oxidative metabolism in response to BRAFV600E-induced senescence was mediated by activation of pyruvate dehydrogenase (PDH), a gatekeeper enzyme that links glycolysis to the TCA cycle and is regulated by phosphorylation by pyruvate dehydrogenase kinase (PDK). Induction of PDH activity in BRAFV600E-induced senescent cells is associated with downregulation of PDK1. This, and the results of another study, suggest that PDK1 inhibitors may synergize with BRAF inhibitors to trigger melanoma cell death [59].
Porchia et al. [60] demonstrated that the compound OSU-03012, a novel PDK1 inhibitor, inhibits thyroid cancer cell proliferation and migration through multiple pathways involved in thyroid cancer progression. However, this compound needs to be validated as an inducer of SL in BRAFV600E-induced thyroid cancer.
MITOCHONDRIAL RESPIRATORY CHAIN DEFECTS AND SYNTHETIC LETHALITY
The genetic basis of cancer progression has been studied extensively. By contrast, the role of mitochondrial genomic mutations in development of thyroid cancer has not. The majority of mitochondrial proteins are encoded by nuclear DNA, with only 37 genes encoded by mitochondrial DNA (mtDNA). These include 13 components of the ETC, 22 transfer RNAs, and two ribosomal RNAs. Several studies show that acquired mtDNA alterations are associated with thyroid cancer. It has been known for over three decades that thyroid tumors contain abnormally high numbers of mitochondria. The majority of the mutations were found in mitochondrial complex I of the respiratory chain, and severe functional defects in complex I activity were observed in thyroid tumor cell lines [61,62].
In addition to the clear genetic alterations in TCA cycle enzymes, various types of cancer display heterogenous mitochondrial OXPHOS function. Therefore, SL strategies based on metabolic reprogramming triggered by these mitochondrial OXPHOS-proficient and -deficient cancers have been proposed. For instance, cancer harboring mutations in TCA cycle enzymes fumarate hydratase (FH) or succinate dehydrogenase (SDH) represent OXPHOS-deficient cancer subtype [63]. Recently, Sun et al. [64] identified metabolic genes that are synthetically lethal with OXPHOS deficiency caused by a defect in FH. Genetic loss-of-function screening found that inhibition of phosphogluconate dehydrogenase (PGD) blocks proliferation of FH mutant cancer cells. It was found that FH mutant cancer cells that exhibit defects in the respiratory chain are sensitive to glycolysis and reductive glutamine carboxylation, which normally lead to activation of respiration [64].
As well as identifying multiple cancers that are highly OXPHOS-proficient, Molina et al. [31] discovered an inhibitor, IACS-010759, which is specifically effective in cancer cells that are deficient in glycolysis. IACS-010759 is an inhibitor of complex I of the mitochondrial ETC. IACS-010759 inhibited the oxygen consumption rate and cell growth by up to 100% in several cancer (pancreatic, lung, ovarian, and breast) cell lines. Glycolysis and OXPHOS co-operate to maintain the cellular energetic balance, and so genetic or pharmacological inhibition of OXPHOS can result in compensatory upregulation of glycolysis to maintain ATP levels. They hypothesized that tumor cells with a reduced capacity for glycolysis would thus be more sensitive to OXPHOS inhibition.
Thyroid cancer cells also showed heterogeneity with respect to OXPHOS function, which can be regulated by genomic alterations, mtDNA mutations, fusion and fission abnormalities, mitophagy, and mitochondrial biogenesis. Application of SL based on OXPHOS function in thyroid cancer requires profiling of mitochondrial function in patients with thyroid cancer.
CONCLUSIONS
Given the high mutational burden of oncogenes such as RET/PTC and BRAFV600E, most differentiated and undifferentiated thyroid cancers carry genetic mutations that provide a therapeutic target. Targeted therapy for BRAFV600E and RET/PTC may not be a successful treatment for patients with radioiodine-refractory and undifferentiated thyroid cancers. Therefore, development of new anticancer drugs based on the concept of SL should facilitate identification of new therapeutic targets, thereby enabling development of new and effective treatment regimens. Moreover, identification of new SL pairs has the potential to drive drug discovery and enable rational testing of drug combinations, with the possibility of translation to thyroid cancer. Translation to the clinic is likely to require collaboration to advance the application of SL drugs to thyroid cancer.
Current areas of interest include exploiting the DNA damage response, metabolic reprogramming, and aberrant tyrosine kinase signaling pathways. At present, relatively few SL pairs have been identified and conclusively validated for thyroid cancer. Furthermore, advances in genome engineering through siRNA and CRISPR/Cas9 technology will provide an effective platform for large-scale screens to identify new SL partners. Future discoveries in the area of SL promise to unveil new therapeutic avenues for effective and personalized treatment of thyroid cancer.
 XML Download
XML Download