

 Citation
Citation Print
Print



INTRODUCTION
Glucose is a primary energy source in mammals including humans and it is thus essential for higher organisms to monitor and control glucose availability within strict physiological ranges. Mammals have a dedicated sensor and effector system for glucose homeostasis in the brain as neurons exclusively use glucose as their energy source, and the brain is the most active glucose consumer. Sensing the blood glucose levels and preventing hypoglycemia is therefore crucial for brain functions such as cognition, memory, and motor functions. The involvement of the brain in glucose regulation was first described in 1854 by French physiologist Claude Bernard who observed a dramatic rise in blood glucose levels when the floor of the fourth ventricle of a rabbit brain was stimulated [1]. In the early 1950s, the French-American scientist Jean Mayer [2] reported that the brain detects glucose fluctuations. In the mid-1960s, experiments using electrophysiological recordings led to the discovery of glucose-sensing neurons and demonstrated their role in maintaining normoglycemia [3–5]. Many studies have since shown that various regions of the hypothalamus and brainstem are involved in the central regulation of glucose metabolism through their interactions with peripheral metabolic organs [6], as depicted in Fig. 1. We here discuss the mechanisms by which the brain regulates glucose metabolism.
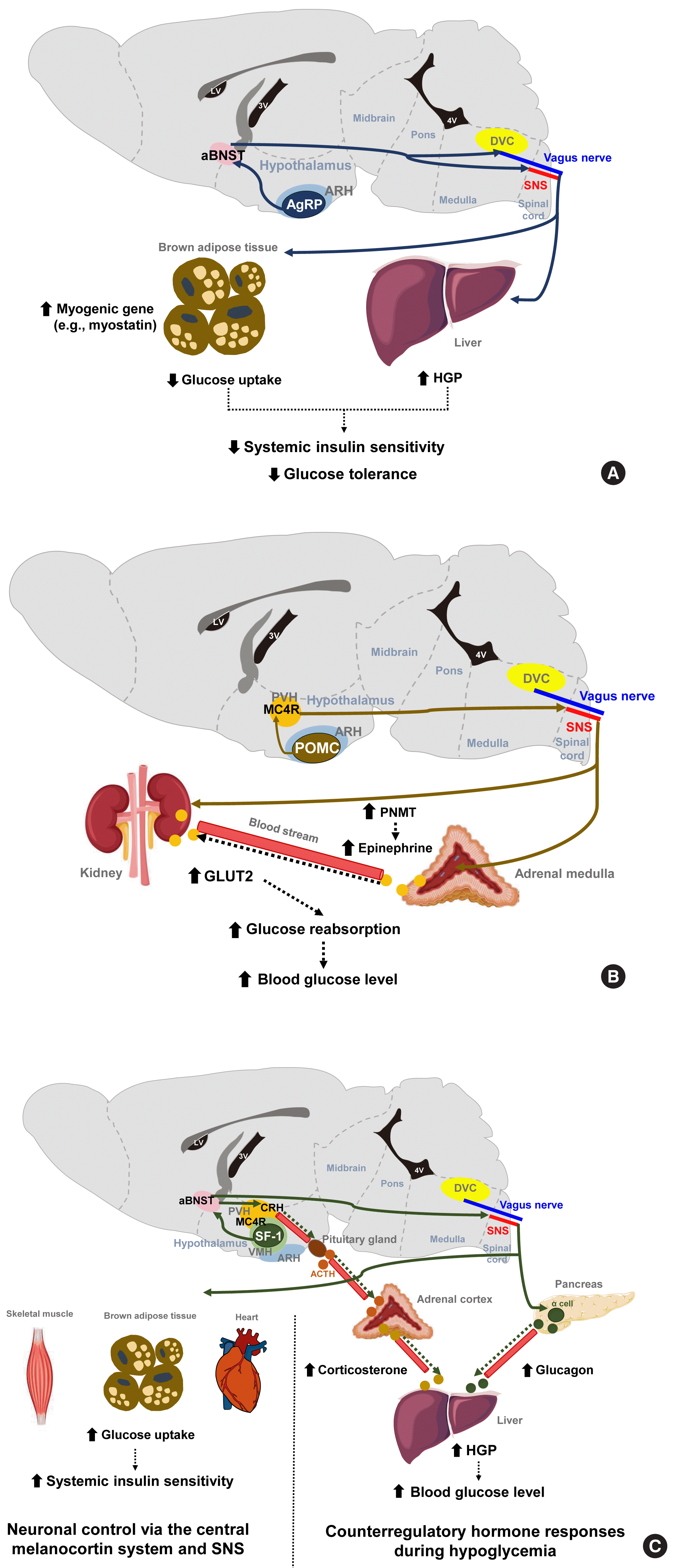 | Fig. 1Representative neurons and their neural circuits that regulate various aspects of glucose metabolism. (A) The optogenetic and chemogenetic stimulation of Agouti-related protein-expressing (AgRP) neurons in the arcuate nucleus of the hypothalamus (ARH) decreases systemic insulin sensitivity and glucose tolerance by reducing glucose uptake into brown adipose tissue. These effects are mediated via neural circuits involving the anterior bed nucleus of the stria terminalis (aBNST) and sympathetic nervous system (SNS) [7]. In addition, activation of ARH AgPP neurons may decrease glucose tolerance by increasing hepatic glucose production (HGP) via the vagal pathway [8]. (B) ARH proopiomelanocortin-expressing (POMC) neurons promote renal glucose reabsorption by increasing glucose transporter 2 (GLUT2) expression in the kidney via paraventricular hypothalamus (PVH) melanocortin receptor-4 (MC4R) neurons and SNS. This regulation occurs directly to the kidney and also indirectly through increased circulating epinephrine by increased phenylethanolamine N-methyltransferase (PNMT) expression in the adrenal medulla [11,12]. (C) During hypoglycemia, steroidogenic factor-1 expressing (SF-1) neurons in the ventromedial hypothalamus (VMH) promote HGP through the secretion of counterregulatory hormones (e.g., glucagon and corticosterone) [14], while they improve insulin sensitivity by increasing glucose uptake in skeletal muscle via the central melanocortin system−SNS [15,16]. ACTH, adrenocorticotropic hormone; DVC, dorsal vagal complex; LV, lateral ventricle; 4V, fourth ventricle.
|
Go to : 
THE NEURONS AND NEURAL CIRCUITS CONTROLLING GLUCOSE HOMEOSTASIS
The hypothalamus has a central function in the neural networks that regulate glucose metabolism. The arcuate nucleus of the hypothalamus (ARH) is located at the bottom of the third ventricle and two neuronal populations in the ARH are considered to be critical for the brain’s regulation of energy balance and glucose metabolism. One of these groups of neurons secretes α-melanocyte stimulating hormone (α-MSH) derived from the precursor peptide proopiomelanocortin (POMC) and the other co-secretes Agouti-related protein (AgRP) and neuropeptide Y (NPY). Regarding energy metabolism, POMC neurons have catabolic actions that are largely through its agonistic actions on the melanocortin-4 receptor (MC4R), whereas AgRP/NPY neurons have anabolic effects that are partly mediated via MC4R antagonism. Recently developed chemogenetic and optogenetic technologies have enabled the control of neuronal activity in specific neuronal populations and neural circuits in live animals in order to study their roles in the control of glucose metabolism.
The stimulation of AgRP neuronal activity using optogenetic and chemogenetic tools decreases the systemic insulin sensitivity and glucose tolerance levels [7]. This effect is partly mediated through the induction of myogenic genes (such as myostatin) and the suppression of glucose uptake in the brown adipose tissue (BAT) and is independent of MC4R antagonism. In addition, AgRP neurons are shown to regulate hepatic glucose production (HGP) [8]. Notably, the neural circuitry involving ARH AgRP neuronal projections to the ventro-lateral part of anterior bed nucleus of the stria terminalis (aBNST) specifically conveys the effects of AgRP stimulation on glucose metabolism (Fig. 1A) [7], whereas their axonal projections to the paraventricular hypothalamus (PVH), the lateral hypothalamus (LH) area, and the dorsomedial part of aBNST control feeding [7].
POMC neurons are shown to mediate the glucose-lowering effects of leptin [9]. In contrast, the acute chemogenetic inhibition of POMC neuronal activity lowers the glycemic level independently of food intake [10]. Moreover, mice that have received ARH-specific POMC ablation show an improved glucose tolerance due to enhanced renal glucose excretion and despite the concomitant induction of obesity and insulin resistance [11]. That same study and another prior investigation reported that ARH POMC neurons promote renal glucose reabsorption via central MC4R signaling, renal sympathetic innervation, and the upregulation of renal expression of type 2 glucose transporter (GLUT2) (Fig. 1B) [11,12].
The ventromedial hypothalamus (VMH) is important for counterregulatory hormone responses and in the recovery from hypoglycemia [13]. When the activity of VMH steroidogenic factor-1 expressing (SF-1) neurons is optogenetically inhibited, animals fail to recover from insulin-induced hypoglycemia owing to impaired counterregulatory hormone responses (reduced glucagon and corticosterone secretion) and decreased HGP [14]. Similar to AgRP neurons, the neural pathway to the aBNST is critical for the regulation of counterregulatory hormone responses by SF-1 neurons [14]. On the other hand, SF-1 neurons also regulate insulin sensitivity in peripheral metabolic organs. The chemical and genetic activation of VMH SF-1 neurons increases systemic insulin sensitivity and glucose uptake in red skeletal muscle, heart and BAT during a hyperinsulinemic-euglycemic clamp [15]. These effects are mediated by central melanocortin signaling and the sympathetic nervous system (Fig. 1C) [16,17]. Consistently, the SF-1 neuron-restricted reexpression of uncoupling protein 2 (UCP2) in generalized UCP2 null mice was reported to activate systemic insulin sensitivity and glucose tolerance by enhancing insulin signaling in the liver, skeletal muscle, and BAT [18].
The LH is known as a feeding center, and its effects are mediated by the inhibitory input from the BNST to the LH glutamatergic neurons [19]. Specific MC4R reexpression in the LH of generalized MC4R-null mice improves glucose tolerance and glycemia in normal chow diet- and high-fat diet (HFD)-fed mice. This glucose-lowering effect of LH MC4R signaling is mediated by upregulation of glucose uptake and thermogenesis in the BAT through the control of BAT sympathetic nerve activity [20]. On the other hand, LH orexin-producing neurons are activated by sweet foods, and they regulate skeletal muscle glucose uptake through VMH orexin receptor-expressing neurons and the sympathetic nervous system [21]. The preoptic area (POA) in the anterior hypothalamus regulates BAT thermogenesis [22]. Given the close link between BAT thermogenesis and glucose tolerance, the involvement of POA neurons in glycemic control needs to be tested.
The dorsal vagal complex (DVC) in the brainstem is crucial for sensing glucose fluctuations and regulating glycemic levels and food intake [23,24]. The DVC includes the nucleus tractus solitarius (NTS), the dorsal motor nucleus of the vagus (DMV), and the area postrema (AP). Meal-related signals from the gastrointestinal tract are relayed to the NTS and the AP through the sensory vagus nerve [25]. Like hypothalamic neurons, NTS neurons produce appetite-regulating peptides such as NPY, POMC, and glucagon-like peptide 1 (GLP-1) [26,27]. The NTS POMC neurons are involved in the short-term regulation of feeding, but the destruction of these neurons neither alters body weight nor glucose metabolism [28]. MC4R is also expressed in the brainstem neurons. Deletion of MC4R in cholinergic sympathetic and parasympathetic preganglionic neurons using Chat-Cre mice impairs glucose homeostasis [29], whereas its reactivation in MC4R null mice attenuates hyperglycemia and insulin resistance through improved hepatic insulin sensitivity. These findings indicate the involvement of brainstem MC4R signaling in the central regulation of glucose metabolism [30]. NTS GABAergic neurons sense hypoglycemia and then help recover from hypoglycemia through DMV projections and the parasympathetic stimulation of glucagon secretion [31]. Conversely, the NTS receives gut-derived, energy-intake signals, which it then transfers to the hypothalamus via the parabrachial nucleus (PBN) (long-loop) or to the DMV (short-loop) to generate satiety [32].
Go to :
GLUCOSE-SENSING MECHANISMS IN THE BRAIN
In the central nervous system (CNS) regulation of glucose metabolism, glucose by itself acts as an important afferent signal to the brain. In this signaling mechanism, blood glucose enters the brain across the blood-brain barrier (BBB), which is mediated by insulin−independent and high-affinity glucose transporter type 1 and 3 (GLUT1, GLUT3) expressed on the endothelial cells. This enables efficient glucose transport into the brain in the normoglycemic range in an insulin-independent manner [33].
Neuronal glucose-sensing
Glucose-sensing neurons are those that can alter their excitability in response to changes in the extracellular glucose levels and can be categorized as glucose-excitatory (GE) and glucose-inhibitory (GI) neurons [34]. GE neurons are excited when the extracellular glucose level rises and vice versa [35]. The molecular basis of glucose-sensing in GE neurons is similar to the classical glucose-sensing mechanism in pancreatic β-cells (Fig. 2A, left panel), although these pathways are not yet fully understood. In GE neurons, glucose enters the neurons via GLUT2, 3, and 4. Upon uptake, glucose is converted to glucose-6-phosphate by glucokinase (GK) and then undergoes glycolysis and mitochondrial oxidation, resulting in adenosine triphosphate (ATP) production. An increased cellular ATP/adenosine diphosphate (ADP) ratio leads to a number of sequential events, i.e., the closure of ATP-dependent potassium (KATP) channels, depolarization of the membrane potential, Ca2+ influx through voltage-dependent calcium channels, and finally the stimulation of neuronal activity and neurotransmitter release [36]. During glucose-sensing by GE neurons, GK activity is essential and is dependent on cellular glucose levels owing to its low affinity for glucose [34]. A wide body of data has illustrated the roles of hypothalamic GLUTs, GK, and KATP channels in glucose-sensing and glycemic control by the CNS [37–41]. For instance, the genetic deletion of Kir6.2, a subunit of the KATP channel, in ARH POMC neurons reduces glucose tolerance [42]. Acute electromagnetic stimulation of GK-expressing neurons in the VMH increases the blood glucose levels by increasing HGP and glucagon levels. In turn, suppressing these neuronal activities curbs the counterregulatory responses to hypoglycemia through opposing actions [43].
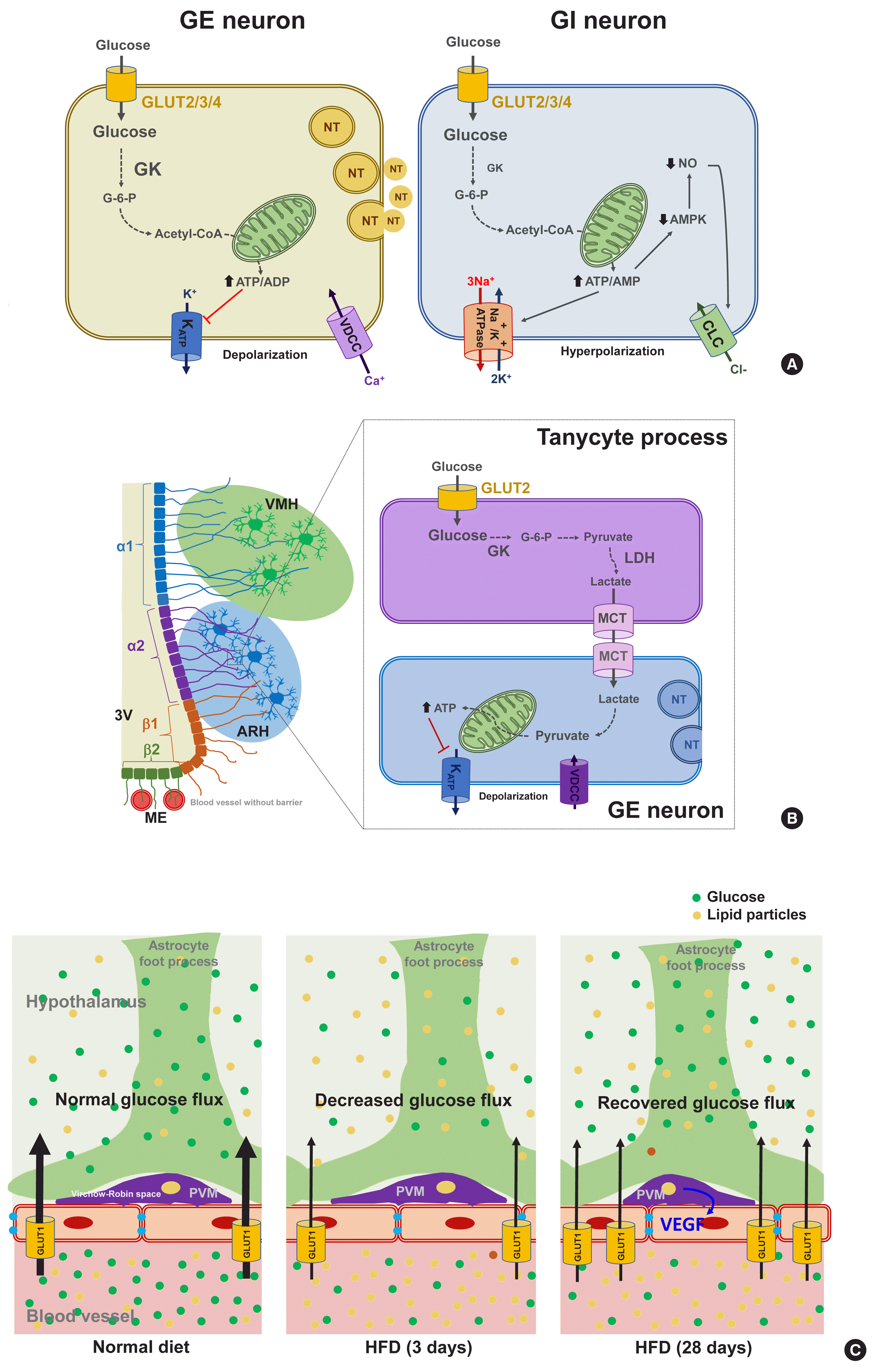 | Fig. 2Molecular mechanisms of glucose-sensing in hypothalamic neurons, tanycytes, and perivascular macrophages. (A) Proposed glucose-sensing mechanisms in hypothalamic neurons. In glucose-excitatory (GE) neurons, glucose enters the neuron through glucose transporter (GLUT) 2/3/4 and increases the intracellular adenosine triphosphate (ATP)/adenosine diphosphate (ADP) ratio, resulting in the closure of ATP-dependent potassium channel (KATP), depolarization of membrane potential, Ca2+ influx through voltage-dependent calcium channel (VDCC), finally releasing neurotransmitter (left panel) [36]. Similarly to GE neuron, glucose enters glucose-inhibitory (GI) neuron through GLUT2/3/4 and increases the intracellular ATP/adenosine monophosphate (AMP) ratio. However, this event leads to inhibition of neuronal activity in GI neurons possibly through stimulation of the Na+/K+-ATPase activity or opening of Cl− channels through suppression of adenosine monophosphate kinase (AMPK) and nitric oxide (NO) production (right panel) [44,46]. Modified from Pozo et al. [33]. (B) Glucose-sensing mechanisms mediated by hypothalamic tanycytes. Glucose enters hypothalamic tanycytes via GLUT2 and converts to lactate via glucokinase (GK) and lactate dehydrogenase (LDH). Tanycyte-produced lactate is released via monocarboxylate transporter (MCT) 1/4 and taken by adjacent neurons via MCT2. In the GE neuron, lactate converts to pyruvate, that produces ATP, and depolarizes neuron action potential via inhibition of KATP channels. Modified from Elizondo-Vega et al. [64]. (C) Perivascular macrophages (PVMs) reside in the Virchow-Robin space regulate glucose flux across the blood-brain barrier (BBB). Glucose flux into the brain decreases upon 3 days-consumption of high-fat diet (HFD) in mice. However, during prolonged HFD feeding (28 days), the PVMs secrete vascular endothelial growth factor (VEGF), which stimulates endothelial GLUT1 expression and thereby restores reduced glucose flux. ARH, arcuate hypothalamus; CLC, chloride channel; G-6-P, glucose-6-phosphate; ME, median eminence; NT, neurotransmitter; 3V, third ventricle; VMH, ventromedial hypothalamus.
|
In contrast to GE neurons, GI neurons are activated when the extracellular glucose level decreases. Relatively little is known however about the underlying mechanism of this. GI neurons also express the same GLUTs and GK as those of GE neurons [34]. However, GK appears to be less essential for glucose-sensing in GI neurons [44]. As a possible mechanism, glucose increases the ATP/adenosine monophosphate (AMP) ratio, which stimulates the Na+/K+-ATPase pump and induces a hyperpolarizing current [45]. Alternatively, glucose may cause the opening of Cl− channels through mechanisms involving adenosine monophosphate-activated protein kinase (AMPK) and nitric oxide (NO), which leads to plasma membrane hyperpolarization (Fig. 2A, right panel) [35,46].
Glucose-sensing neurons are mainly distributed in the hypothalamic, corticolimbic, and brainstem areas, also known as critical body weight control areas. As the ARH is adjacent to the circumventricular organ median eminence (ME), ARH neurons are the first to sense the changes in blood glucose. Many studies have indicated that POMC neurons are GE, and about 40% of AgRP/NPY neurons are GI [34]. GE neurons are also found in the VMH and the PVH, which are critically involved in satiety generation [47]. In contrast, GI neurons are distributed in the LH, which has long been known as a hunger center [35,48].
Both GE and GI neurons are also present in the brainstem areas, including the DMV, NTS, and PBN [49–51], whereas the PBN mostly contains GI neurons [52], which is consistent with its role in the adaptative responses to hypoglycemia. In further support of these findings, the local induction of glucoprivation in these aforementioned brainstem areas using a 5-Thio-D-glucose injection triggers hyperglycemic and hyperphagic responses in rats [53]. Glucose-sensing neurons are also found in the limbic regions related to the rewarding such as the nucleus accumbens (NAc) and amygdala [54]. The NAc and amygdala contain both GE and GI neurons. The reasons why neurons in the reward system need to sense glucose remain to be resolved.
Non-neuronal glucose-sensing
The mammalian brain comprises both neurons and non-neuronal cells (astrocytes, microglia, oligodendrocytes, nerve/glial antigen 2 [NG2] glia, endothelial cells, pericytes, ependymal cells, and tanycytes). Recent evidence indicates an involvement of non-neuronal cells in brain glucose-sensing. Astrocytes are abundant glial cells that support neuronal functions in many ways. As astrocytes are located at the interface between blood vessels and neurons, they are in a prime location for controlling the glucose flux into the CNS. The foot processes of astrocytes that ensheath the brain microvessels, highly express GLUT1. Blood glucose enters the astrocytes mainly through GLUT1 and then is oxidized in the mitochondria, stored as glycogen, or converted to lactate and secreted. Lactate is an alternative energy source for neurons. Indeed, neurons are believed to be metabolically and functionally coupled with astrocytes through lactate, which is known as the astrocyte-neuron lactate shuttle hypothesis [55].
Interestingly, persistent hyperglycemia in streptozotocin-induced diabetic rats was reported to reduce the astroglial GLUT1 expression level, and to impair elevation in the hypothalamic glucose concentrations, and HGP suppression after glucose loading [56]. These findings suggest that GLUT1-mediated glial glucose uptake is important for brain glucose-sensing and for the regulation of glucose metabolism. On the other hand, a generalized depletion of GLUT2 is known to impair glucagon secretion during hypoglycemia, and to be rescued by the selective reexpression of GLUT2 in glial fibrillary acidic protein (GFAP)-expressing astroglial cells, but not in neurons [38]. This evidence indicates the critical role of astroglial GLUT2 in sensing hypoglycemia and regulating glucagon secretion.
Tanycytes are specialized ependymal cells surrounding the third cerebroventricle [57] and have four subtypes: α1, α2, β1, and β2. They are polarized cells as their cell body faces the cerebroventricular side, and their long processes project and contact the neurons in the VMH (α1) and ARH (α2 and β1), or the fenestrated blood vessels in the ME (β2) [58,59]. Thanks to topological characteristics, the tanycytes mediate material transport between the cerebroventricle and the hypothalamic parenchyma (α1, α2, β1) and between the cerebroventricle and blood vessels (β2). For instance, tanycytes have been shown to mediate leptin transport from the blood to the hypothalamus [60]. These cells also express the glucose-sensing molecules GLUT2, GK, and KATP channels [61] and may sense glucose in the cerebrospinal fluid (CSF) which is derived from the blood across the blood–CSF barrier. As indicated in Fig. 2B, tanycytes may play a role in glucose uptake from the CSF via GLUT2 and then release lactate through monocarboxylate transporter. Tanycyte-derived lactate may activate or inhibit the neuronal electrical activity that contacts the processes of these cells, as demonstrated in vitro in a prior study [62]. The selective destruction of tanycytes through the intracerebroventricular injection of alloxan inhibits the counterregulatory responses to hypoglycemia without damaging ARH neurons [63]. These findings support the involvement of tanycytes in hypothalamic glucose-sensing [64], as depicted in Fig. 2B.
Perivascular macrophages (PVMs) are located in the Virchow-Robin space, a fluid−filled region that surrounds the small arteries in the brain. Given the location of this space at the interface of blood vessels and the brain parenchyma, these cells may sense and regulate glucose flux from the circulation to the brain. Under conditions of a reduced glucose flux, i.e., HFD consumption, PVMs secrete vascular endothelial growth factor (VEGF) which, in turn, acts on the endothelial cells to restore the glucose flux by increasing GLUT1 expression (Fig. 2C) [65]. This mechanism appears to be critical for adequate glucose supply to the CNS neurons and maintenance of cognitive functions in HFD-fed conditions. However, whether this may also be important for controlling glucose metabolism has not been tested yet. Microglia are macrophage-like cells that are evenly distributed throughout the brain parenchyma and execute multiple homeostatic roles and innate immune functions. A recent study has shown that microglia, especially those in proximity to the glucose-sensing NPY neurons, are activated during hypoglycemia. Inhibiting microglial activation using minocycline enhances the counterregulatory hormone responses to hypoglycemia [66], and thus microglial activation around NPY neurons may play a role in the hypoglycemia unawareness induced by repeated exposure to hypoglycemia. These findings imply that microglia may sense fluctuations in nutrient availability and regulate neuronal adaptive responses. Evidence for the contribution of hypothalamic immune cells to glucose homeostasis is beginning to be reported. Therefore, future studies are necessary to reveal how hypothalamic immune cells affect glucose metabolism.
Go to :
CONTRIBUTION OF CENTRAL INSULIN SIGNALING TO SYSTEMIC GLUCOSE METABOLISM
Insulin is the most important hormone in glucose regulation and is generally thought to regulate glucose metabolism via its direct actions on peripheral metabolic organs. However, insulin receptor (IR) and its downstream signaling molecules, insulin receptor substrate 1 and 2 (IRS1/IRS2), are expressed in brain regions including the hypothalamus [67]. Moreover, neuron-specific IR knockout (NIRKO) mice exhibit mild obesity and insulin resistance [68]. Brain-specific IRS2 knockout mice also display overweight, hyperinsulinemia, and glucose intolerance [69]. These findings support the notion that insulin also regulates glucose metabolism and body weight through its actions in the brain.
Several lines of evidence suggest that central insulin signaling improves peripheral glucose metabolism. For example, insulin-induced suppression of HGP was reported to be attenuated when the brain IR and its downstream signaling were inhibited, regardless of the circulating insulin levels [70]. Moreover, insulin infusion into the cerebroventricle was shown to suppress HGP and this effect was transmitted to the liver via hepatic vagal innervation and the α7-nicotinic acetylcholine receptor (Fig. 3A) [71]. Thus, insulin suppresses HGP through the CNS and via vagus nerve-mediated mechanisms. ARH AgRP neurons mediate this central effect of insulin on HGP as the restoration of IR in these neurons in generalized IR null mice rescues the ability of insulin to suppress HGP [8]. In AgRP neurons, insulin inhibits neuronal activity through phosphatidyl-inositol-3 kinase (PI3K)-dependent mechanisms and via the opening of KATP channels (Fig. 3A) [72]. Other important insulin-target neurons with respect to HGP control are located in the brainstem DVC. Whereas PI3K mediates insulin signaling in the hypothalamic neurons, extracellular signal-regulated kinase (ERK) mediates insulin signaling in the DMV neurons through the direct phosphorylation and activation of KATP channels (Fig. 3A) [73]. With regard to hepatic mechanisms, central IR signaling activates hepatic signal transducer and activator of transcription 3 (STAT3) signaling through increased interleukin-6 (IL-6) production, which leads to a reduced expression of gluconeogenic genes (Pepck, G6Pase) and HGP [74]. Hence, the actions of central insulin on the HGP levels is believed to be mediated by neural KATP channel−vagus−hepatic IL-6/STAT3 signaling.
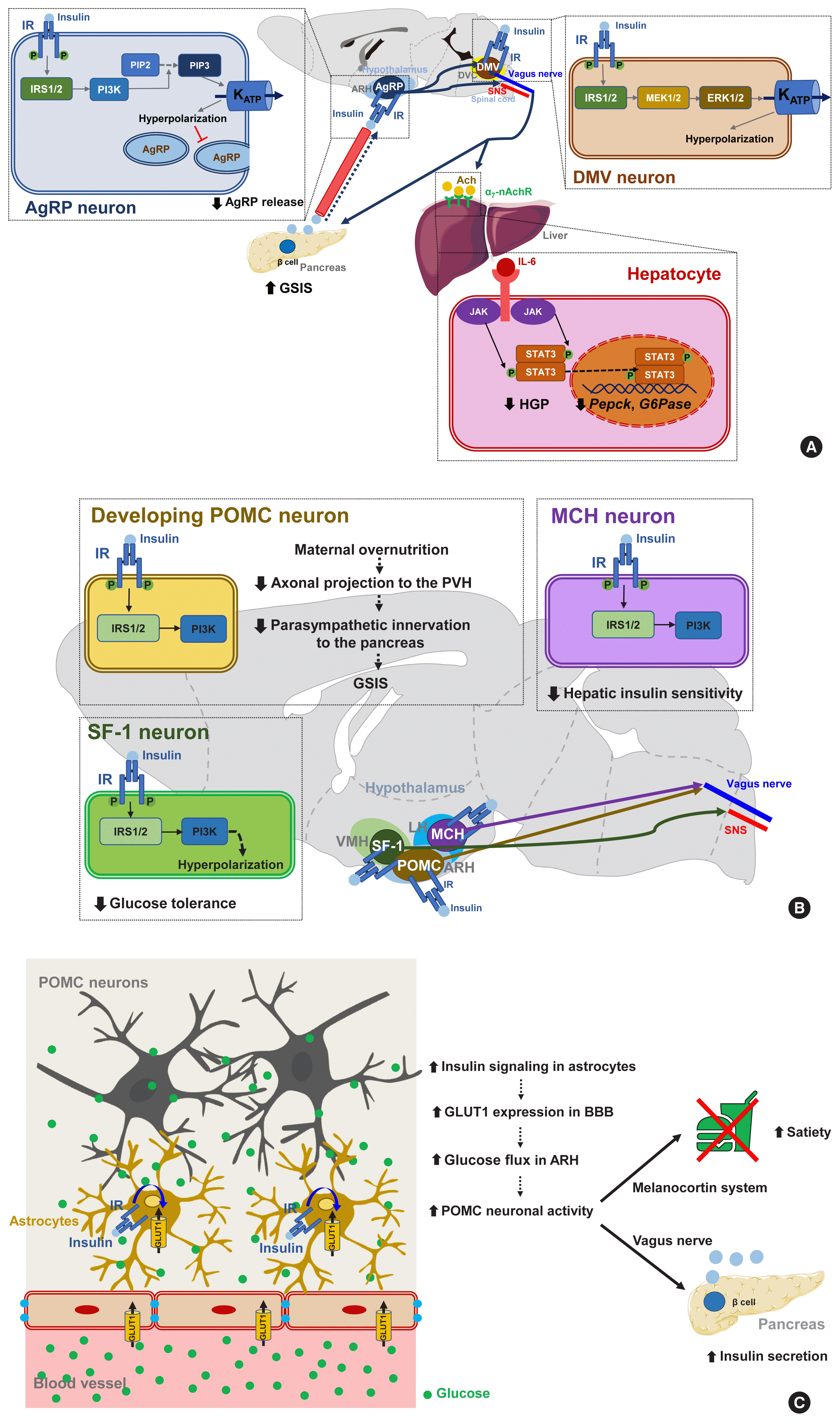 | Fig. 3Insulin signaling in hypothalamic neurons and astrocytes regulates systemic glucose metabolism. (A) Insulin signaling in hypothalamic Agouti-related protein (AgRP) and dorsal motor nucleus of the vagus (DMV) neurons. In these neurons, insulin inhibits neuronal activity through the phosphatidyl-inositol-3 kinase (PI3K)- and extracellular signal-regulated kinase (ERK)-dependent opening of ATP-dependent potassium (KATP) channels [72,73]. Activated insulin signaling in AgRP neurons inhibits hepatic glucose production (HGP) [74] and stimulates glucose-stimulated insulin secretion (GSIS) from the pancreatic β-cells [78]. (B) Overnutrition causes hyperinsulinemia, a condition that adversely affects glucose homeostasis through its effects on specific hypothalamic neurons [83]. The activation of insulin signaling in lateral hypothalamus (LH) melanin-concentrating hormone (MCH) neurons decreases hepatic insulin sensitivity [85]. Insulin signaling in ventromedial hypothalamus (VMH) steroidogenic factor-1 expressing (SF-1) neurons promotes obesity and glucose dysregulation by inhibiting SF-1 neuronal activity [86]. Enhanced insulin signaling in developing proopiomelanocortin (POMC) neurons suppresses GSIS by reducing POMC axonal projections to the paraventricular hypothalamus (PVH) and the parasympathetic innervation of pancreatic islets [84]. (C) Astrocytic insulin signaling stimulates the glucose flux into the hypothalamus by increasing blood-brain barrier (BBB) glucose transporter 1 (GLUT1) expression. This helps POMC neurons to generate satiety and stimulate GSIS [87]. Modified from Garcia-Caceres et al. [87]. α7-nAchR, α7 nicotinic acetylcholine receptor; Ach, acetylcholine; ARH, arcuate nucleus of the hypothalamus; DVC, dorsal vagal complex; IL-6, interleukin-6; IR, insulin receptor; IRS, insulin receptor substrate; JAK, Janus kinase; MEK, mitogen-activated protein kinase; PIP2, phosphatidyl-inositol diphosphate; PIP3, phosphatidyl-inositol triphosphate; SNS, sympathetic nervous system; STAT3, signal transducer and activator of transcription 3.
|
In addition to HGP regulation, central insulin signaling may regulate systemic insulin sensitivity. In humans, intranasal insulin administration, which delivers insulin to the brain, increases peripheral fat mobilization, thermogenesis, and peripheral insulin sensitivity [75]. This insulin-mediated thermogenic regulation may underlie diet-induced thermogenesis. Consistently, the artificial activation of AgRP neurons in mice using optogenetic techniques inhibits insulin-stimulated glucose uptake in the BAT by increasing myostatin expression (Fig. 3A) [7]. Given the inhibitory effect of insulin on AgRP neuronal activity, it has been speculated that insulin may stimulate glucose uptake by the BAT through AgRP inhibition. Meanwhile, insulin inhibits lipolysis in the adipose tissue via central mechanisms and, via a process which is mediated by POMC neuronal insulin signaling, dampens the sympathetic flow to the adipose tissues [76]. As skeletal muscle is an important site of glucose processing, it is possible that central insulin signaling may regulate glucose metabolism in skeletal muscles. Indeed, a prior study has found that intracerebroventricular insulin infusion increases muscle glycogen synthesis [77], although the details of the mechanisms and neuronal circuits underlying this effect remain to be revealed.
An additional regulatory aspect of insulin is that it acts on the brain to regulate its own secretion. Hypothalamic GK-expressing glucose-sensing neurons display multisynaptic connections to the pancreas via brainstem DMV neurons and vagal innervation [78]. The ablation of hypothalamic GLUT4-expressing neurons leads to a reduction in insulin secretion [79]. Brain insulin signaling is also required for counterregulatory responses to hypoglycemia [80]. NIRKO mice exhibit a blunted sympathoadrenal response to hypoglycemia due to reduced glucose-sensing in their hypothalamic neurons [81,82].
It is noteworthy that insulin signaling in certain subsets of neurons can cause adverse metabolic outcomes under overnutrition conditions (Fig. 3B) [83]. Insulin signaling in POMC neurons may have a negative impact on peripheral glucose metabolism as selective IR expression in POMC neurons in whole-body IR knockout mice aggravates insulin resistance and increases HGP. Maternal overnutrition during lactation predisposes offspring to metabolism disorders in adulthood [84]. It has been further shown that maternal HFD feeding reduces the axonal projections of ARH POMC neurons to the posterior part of the PVH and also suppresses the parasympathetic innervation of pancreatic islets, leading to reduced glucose-stimulated insulin secretion (GSIS) and glucose intolerance (Fig. 3B). Interestingly, POMC-specific IR deletion in mouse pups rescues maternal overnutrition-induced disturbances in POMC neuron-to-islet circuit formation and glucose metabolism. Therefore, maternal overnutrition in the mouse may overactivate insulin signaling in POMC neurons in pups, which possibly interrupts the neural circuit organization from hypothalamic POMC neurons to pancreatic islets and causes a long-lived defect in glucose metabolism in their progeny.
Similarly to the above results, insulin increases the electrical activity of melanin-concentrating hormone (MCH)-producing neurons in the LH, which leads to impaired glucose homeostasis through the control of hepatic insulin sensitivity and HGP (Fig. 3B) [85]. Activated insulin signaling, i.e., increased PI3K activity, is observed in the MCH neurons of HFD-fed mice [85]. Thus, insulin signaling in MCH neurons may mediate the adverse metabolic outcomes of overnutrition-associated hyperinsulinemia. On the other hand, insulin inhibits VMH SF-1 neuronal activity through a similar mechanism to AgRP neurons. Interestingly, SF-1 neuron-specific IR deletion curbs HFD-induced obesity, hyperinsulinemia, and glucose intolerance [86]. This result suggests that insulin acts on VMH SF-1 neurons to promote obesity and glucose dysregulation (Fig. 3B).
A recent study has corroborated the importance of astrocyte insulin signaling in the brain regulation of glucose metabolism (Fig. 3C). In that report, the genetic ablation of IR in astrocytes was found to lead to impaired metabolic responses to blood glucose fluctuations [87]. Mice lacking astrocytic IRs show reduced satiety and lower insulin secretion and develop higher blood glucose levels following peritoneal glucose injection. This impairment is related to a reduced glucose flux to the hypothalamus due to lower astrocytic GLUT1 expression. Moreover, these mice have reduced hyperphagia upon exposure to hypoglycemia [87]. These results indicate that astrocyte insulin signaling critically mediates glucose-sensing and regulation by the CNS via the control of glucose transport across the BBB.
Go to :
OTHER HORMONAL SIGNALING MECHANISMS INVOLVED IN BRAIN REGULATION OF GLUCOSE METABOLISM
Leptin is an adipocyte-derived hormone that regulates not only body weight but also glucose metabolism by acting on the brain. A leptin infusion of the cerebroventricle improves hyperglycemia in rats with insulin-deficient diabetes. The glucose-lowering effect of leptin is due to reduced HGP and increased insulin-independent glucose uptake into the brain, skeletal muscle, and BAT (Fig. 4A). The first-order neurons mediating leptin actions are distributed in the ARH. In leptin receptor-null mice, the reconstitution of leptin receptors, specifically in ARH POMC neurons, normalizes the blood glucose levels and these effects are independent of food intake and body weights. Moreover, mice with double knockout of insulin and leptin receptors in POMC neurons cannot suppress HGP during the hyperinsulinemic-euglycemic clamp, and these animals develop systemic insulin resistance and glucose intolerance [88]. Hence, ARH POMC neurons seem to mediate the modulation of peripheral glucose metabolism by leptin. Several lines of evidence suggest that leptin also regulates skeletal muscle glucose metabolism via the VMH neurons. Local leptin injections into the VMH increase glucose uptake in mouse skeletal muscle and improve glucose tolerance independently of the circulating insulin levels [89,90]. These effects are abolished when the sympathetic nervous system and central melanocortin receptor signaling are inhibited [16]. Central leptin injections enhance insulin-stimulated Akt phosphorylation and also activate AMPK signaling in skeletal muscle [91,92]. Both signaling events are expected to increase glucose uptake in skeletal muscle.
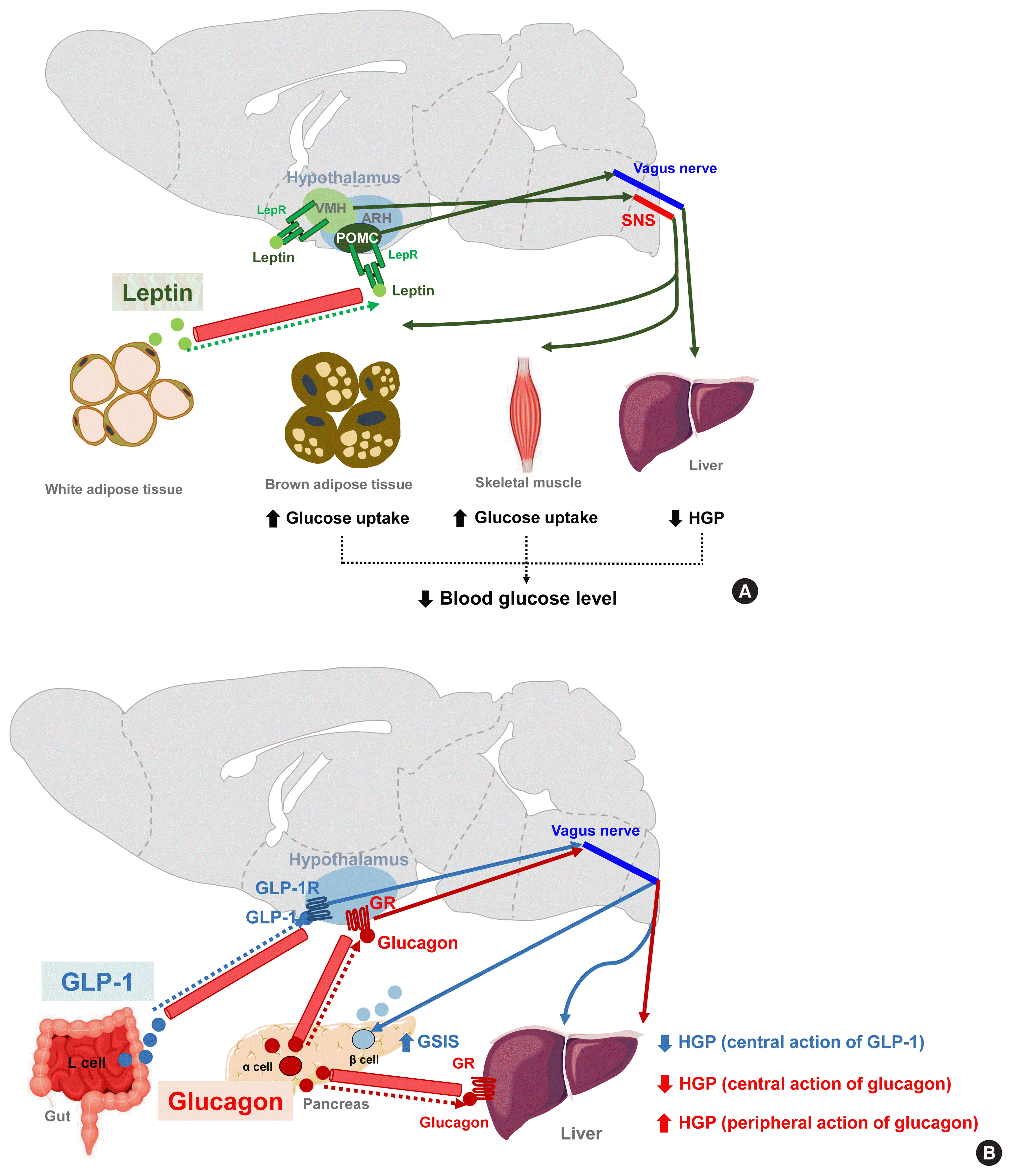 | Fig. 4Hormonal (leptin, glucagon-like peptide 1 [GLP-1], and glucagon) signaling modulates peripheral glucose metabolism via central actions. (A) Leptin signaling in both the arcuate nucleus of the hypothalamus (ARH) proopiomelanocortin-expressing neuron (POMC) and the ventromedial hypothalamus (VMH) neurons leads to decreased hepatic glucose production (HGP) and increased insulin-independent glucose uptake in the brain, skeletal muscle, and brown adipose tissue (BAT) [88]. By acting on VMH neurons, leptin stimulates skeletal muscle glucose uptake via the central melanocortin receptor−sympathetic nervous system (SNS)−skeletal muscle AMP-activated protein kinase (AMPK) signaling mechanism [16,91,92]. (B) GLP-1 increases glucose-stimulated insulin secretion (GSIS) but inhibits HGP through GLP-1 receptor signaling in the ARH [93]. Glucagon enhances HGP via a direct action on hepatocytes whereas it acts on the hypothalamus to inhibit HGP [94]. AC, adenylyl cyclase; ACC, acetyl-CoA carboxylase; α-AR, α-adrenergic receptor; α2-AMPK, α2-catalytic subunit of adenosine monophosphate kinase; β2-AR, β2-adrenergic receptor; GLP-1R, GLP-1 receptor; I3K, phosphatidyl-inositol-3 kinase; GR, glucagon receptor; IRS, insulin receptor substrate; KATP, ATP-dependent potassium channel; LepR, leptin receptor; PKA, protein kinase A.
|
GLP-1 is a gut-derived hormone with an incretin effect. In addition to its direct effects on pancreatic islets, GLP-1 augments GSIS and suppresses HGP through GLP-1 receptor signaling in the ARH (Fig. 4B) [93]. Glucagon, secreted by pancreatic α-cells, is thought to elevate blood glucose levels by acting on the hepatocytes and increasing HGP. In contrast to this peripheral action, the activation of hypothalamic glucagon receptor signaling inhibits HGP through a KATP channel-dependent mechanism (Fig. 4B) [94]. These opposing effects of central glucagon signaling on HGP may constitute a negative feedback loop that self-limits glucagon-stimulated HGP and fine-tunes the regulation of HGP by glucagon.
Go to :
OBESITY-INDUCED HYPOTHALAMIC INFLAMMATION AND ITS POSSIBLE IMPACTS ON GLUCOSE HOMEOSTASIS
Obesity is a major risk factor for type 2 diabetes and, along with the intake of saturated fatty acids, can disrupt insulin signaling and glucose metabolism in peripheral metabolic organs. Several mechanisms such as the activation of inflammation signaling, endoplasmic reticulum stress, mitochondrial dysfunction, and accumulation of toxic lipid metabolites have been suggested to bridge obesity and metabolic disorders. Similar pathological events may also occur in the brain areas that control glucose metabolism [95].
The repeated consumption of fat-rich diets activates and accumulates microglia, which are brain-resident innate immune cells, in the ARH. Activated microglia secrete proinflammatory cytokines and elicit inflammatory signaling pathways in adjacent neurons. A considerable amount of data has now corroborated the evidence that activated inflammatory signaling hampers leptin and insulin signal transduction in hypothalamic neurons and that these signaling changes disturb the hormone-mediated hypothalamic regulation of glucose metabolism [96].
The accumulation of GFAP-expressing reactive astrocytes, so-called astrogliosis, is another cardinal feature of obesity-associated hypothalamic changes [97]. Given the critical roles of astrocytes in hypothalamic glucose-sensing, astrogliosis may possibly contribute to glucose dysregulation in obese animals and humans. Indeed, the inhibition of nuclear factor kappa B (NF-κB) signaling in astrocytes significantly attenuates obesity progression and glucose dysmetabolism during HFD feeding [98]. However, it is unclear whether improved glucose metabolism in these mice is secondary to or independent of weight loss.
Hypothalamic PVMs, which reside between the hypothalamic parenchyma and blood vessels, may contribute to obesity-induced alteration in glucose metabolism. As mentioned in the section on non-neuronal glucose-sensing, PVMs help to maintain glucose transport into the hypothalamus through VEGF secretion under the HFD-fed condition [83]. On the other hand, chronic HFD consumption inflames PVMs to secrete proinflammatory cytokines and boost hypothalamic inflammatory responses to fatty acids. Activated PVMs also release reactive oxygen species such as nitric oxide (NO) [99]. Enhanced NO production in PVMs may increase the permeability of BBB, leading to increased fatty acid flux in the hypothalamus [99]. As a result, fatty acids-induced hypothalamic inflammation and disturbances in hypothalamus-mediated glucose regulation may be accelerated. Supporting this notion, a blockade of inducible NO synthase (iNOS) in hypothalamic PVMs significantly improves systemic insulin resistance, the homeostatic model assessment of insulin resistance (HOMA-IR) index, and glucose intolerance in HFD-fed obese mice [99]. These findings suggest that iNOS signaling in the hypothalamic PVMs may regulate systemic glucose metabolism and insulin resistance via as yet unclear mechanisms. Collectively, overnutrition-induced glucose intolerance and insulin resistance clearly involve central mechanisms, as depicted in Fig. 5.
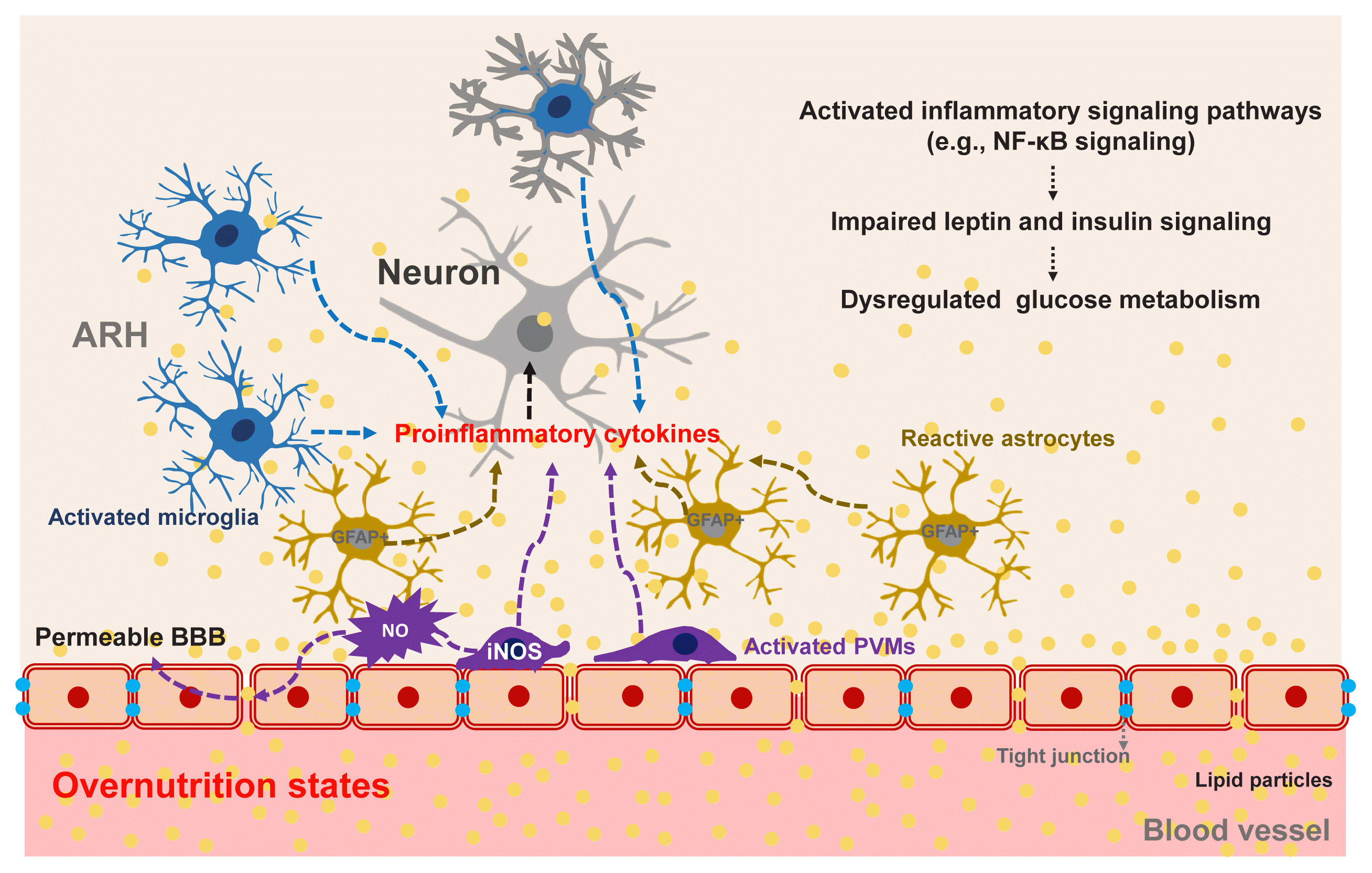 | Fig. 5Obesity-induced hypothalamic inflammation and its possible impacts on glucose homeostasis. Overnutrition causes inflammation in the arcuate nucleus of the hypothalamus (ARH), which is mediated by activated microglia, perivascular macrophages (PVMs), and reactive astrocytes [96–99]. Upon persistent exposure to fat-rich diets, these glial cells secrete proinflammatory cytokines and reactive oxygen species (ROS), thereby activating inflammatory signaling pathways and disrupting leptin and insulin signaling in adjacent neurons [99]. Activation of inducible nitric oxide synthase (iNOS) in hypothalamic PVMs increases the permeability of the blood-brain barrier (BBB), which results in increased fatty acid flux into the ARH and accelerated fatty acid-induced hypothalamic inflammation [99]. NF-κB, nuclear factor kappa B; GFAP, glial fibrillary acidic protein; NO, nitric oxide.
|
Go to :
CONCLUSIONS
This review highlights the robust evidence that the brain plays a vital role in the homeostatic regulation of glucose metabolism. The brain has highly organized machinery, differentiated by cell type and region (hypothalamic and brainstem), that can detect changes in systemic glucose metabolism. The hypothalamic and brainstem neurons also sense the peripheral metabolic state through hormones such as insulin, leptin, GLP-1, etc. In addition to neurons that have established roles in the brain, recent evidence has now revealed an essential role of the glial cells in the regulation of glucose metabolism. To achieve glucose homeostasis, the brain modulates peripheral organ glucose metabolism in many different ways, i.e., control of glucose flux in the liver and skeletal muscle, insulin/glucagon secretion in the endocrine pancreas, glucose reabsorption/secretion in the kidney, and as yet unidentified processes. The specific neurons and their circuits that are involved in each aspect of peripheral organ glucose metabolism are beginning to be revealed. A better understanding of the neuronal mechanisms of glucose homeostasis will likely lead to the discovery of novel therapeutic targets for combating hyperglycemia and hypoglycemia.
Go to :
 XML Download
XML Download