

 Citation
Citation Print
Print



INTRODUCTION
Fatty liver disease, also known as hepatic steatosis, is characterized by excessive fat build-up in the liver and is closely associated with diabetes, hypertension, cardiovascular diseases, and liver cancer [1,2]. Nonalcoholic fatty liver disease (NAFLD) is involved in the control of glucose and lipid metabolism and ranges from simple fatty liver to nonalcoholic steatohepatitis (NASH) [3,4]. Lipid metabolism in the hepatocytes maintains homeostasis by balancing the uptake of circulating fatty acids, fatty acid oxidation (in the mitochondria, peroxisomes, and cytochromes), de novo lipogenesis, and very-low-density lipoprotein secretion [5]. Recently, the protective effect of anti-diabetic drugs, such as dipeptidyl peptidase 4 (DPP-4) inhibitors, sodium glucose cotransporter 2 inhibitors, and glucagon-like peptide-1 receptor agonists (GLP-1RAs) on NAFLD/NASH in humans has been reported [6].
Glucagon-like peptide-1 (GLP-1) is an incretin hormone released from the gut in response to the presence of nutrients. GLP-1 has several beneficial functions, such as stimulating insulin secretion and beta-cell proliferation in the pancreas, promoting glucose production and glucose uptake in the liver, increasing glucose uptake and glycogen production in the skeletal muscle, decreasing food intake, and delaying gastric emptying [7,8]. However, GLP-1 has a short half-life (1 to 1.5 minutes) due to degradation by the DPP-4 enzyme [9]. GLP-1RAs, which are peptides based on either exendin-4 or human GLP-1, resist the activity of DPP-4. Dulaglutide is a long acting GLP-1RA and contains a human GLP-1 backbone [10]. Dulaglutide reduces body weight and A1C levels in patients with type 2 diabetes [11], as well as reduces alanine aminotransferase and body mass index in NAFLD patients with type 2 diabetes [12,13]. However, the regulatory mechanism by which dulaglutide induces its beneficial effects in hepatic steatosis remains to be elucidated.
The family with sequence similarity 3 (FAM3) gene family consists of four members, FAM3A, FAMB, FAM3C, and FAM3D, which play key roles in regulating glucose and lipid metabolism. FAM3A is ubiquitously expressed in all tissues including the liver, spleen, and adipose [14]. The expression of FAM3A is reduced in the liver and adipose tissue of obese mice. While FAM3A overexpression in db/db mice improves glucose intolerance, hyperglycemia, and insulin resistance, it attenuates gluconeogenesis and lipogenesis [15–17]. In addition, FAM3A overexpression increases the expression of hepatic adipoR1, which is involved in lipid oxidation by increasing uncoupling protein 2 (UCP2) and adenosine monophosphate (AMP)-activated protein kinase (AMPK) expression [15,18].
In the present study, we investigated the protective effects of a novel GLP-1RA, dulaglutide, in PA-induced hepatic steatosis and examined the functional roles and the molecular mechanisms of dulaglutide-induced increase in FAM3A in palmitic acid (PA)-treated HepG2 cells.
METHODS
Cell culture
The human hepatoma cell line HepG2 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). PA (Sigma-Aldrich, St. Louis, MO, USA) was used to induce hepatic steatosis in cultured hepatocytes. To examine the protective effect of dulaglutide on PA-induced steatosis, cells were pretreated with 400 μM PA for 24 hours, followed by treatment with or without 100 nM dulaglutide (MedChemExpress, Monmouth Junction, NJ, USA) for 24 hours. To investigate whether the beneficial effects of dulaglutide in hepatic steatosis are mediated by GLP-1R, cells were pretreated with 400 μM PA for 24 hours, followed by treatment with or without 100 nM dulaglutide and 100 nM exendin (9–39) (Sigma-Aldrich) for 24 hours. The cells were cultured in an incubator at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Triglyceride quantification and Oil red O staining
To measure total intracellular triglyceride (TG) content, lipids were extracted from the cultured cells and total TG content was determined by enzymatic assays (TR0100, Sigma-Aldrich) and normalized to protein concentration. The protein concentration was measured using the Bradford assay (Bio-Rad, Munich, Germany). HepG2 cells were seeded in 6-well plates and stained using the Oil red O method. Briefly, after treatment, cells were washed three times with phosphate-buffered saline (PBS) and fixed for 30 minutes with 10% formalin. After fixation, cells were washed again with PBS, stained with Oil-Red O (Sigma-Aldrich) for 1 hour, and then rinsed with distilled water. The cells were observed under a light microscope (Olympus, Tokyo, Japan) with a 400× magnification and images were photographed. For quantitative analysis of cellular lipid, 1 mL isopropanol (Sigma-Aldrich) was added to the stained culture plate and absorbance of the extracted dye was recorded at 540 nm.
Knockdown by siRNA
To silence the target protein, small interfering RNA (siRNA) duplexes targeting FAM3A and negative control siRNA were purchased from Bioneer (Daejeon, Korea), and the cells were transiently transfected with synthetic siRNAs using the Lipofectamine RNAi MAX transfection reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. After 24 hours of transfection, the cells were pretreated with 400 μM PA for 24 hours, followed by treatment with or without 100 nM dulaglutide for 24 hours. The cells were then harvested for further analysis, and knockdown efficiency of the siRNAs was determined by Western blotting.
Quantitative real time polymerase chain reaction
Total RNA was isolated from cultured HepG2 cells using the TRIzol reagent (Invitrogen). The RNA was reverse-transcribed using the High-Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. Gene expression analysis was performed by quantitative real time polymerase chain reaction using LightCycler 480 SYBR Green (Roche, Lewis, UK). Primer sequences used for the detection of genes are shown in Supplemental Table S1. The expression levels of the target genes were normalized to the housekeeping control gene (18S rRNA) and relative quantification of the target gene mRNA was performed using the 2−ΔΔCt method.
Western blotting
Total protein extracts were isolated from the HepG2 cells. The cells were lysed with RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) containing the Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Rockford, IL, USA) on ice. Then, protein concentrations were measured and equal amounts of the protein samples (15 μg) were dissolved in 4× lithium dodecyl sulfate sample buffer and 10× reducing sample agent (Invitrogen) and denatured at 95°C for 10 minutes. The samples were loaded on a 4% to 12% Bis-Tris NuPAGE gel (Thermo Fisher Scientific). The separated proteins were transferred from the gel to a polyvinylidene difluoride membrane by using the iBlot2 PVDF stack (Invitrogen). The membranes were blocked with 5% bovine serum albumin in tris-buffered saline and tween 20 (TBST) for 1 hour at room temperature and probed at 4°C overnight with the following primary antibodies: anti-sirtuin 1 (SIRT1) (sc-15404, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), anti-phospho-AMPK (#2531, Cell Signaling Technology), anti-AMPK (#2532, Cell Signaling Technology), anti-perilipin 1 (PLIN1) (sc-67164, Santa Cruz Biotechnology Inc.), anti-adipose differentiation-related protein (ADFP) (ab52355, Abcam, Cambridge, MA, USA), anti-adipose triglyceride lipase (ATGL) (#2138, Cell Signaling Technology), anti-phospho-hormone-sensitive lipase (p-HSL) (#4126, Cell Signaling Technology), anti-phospho-acetyl-CoA carboxylase (p-ACC) (#3661, Cell Signaling Technology), anti-peroxisome proliferator-activated receptor alpha (PPARα) (ab8934, Abcam), anti-FAM3A (SAB1102488, Sigma-Aldrich), anti-GLP-1R (sc-66911, Santa Cruz Biotechnology Inc.), anti-Ca2+/calmodulin-dependent protein kinase kinaseβ (CaMKKβ) (sc-271674, Santa Cruz Biotechnology Inc.), and anti-β-actin (#4967, Cell Signaling Technology). The membranes were incubated for 1 hour at room temperature with horseradish peroxidase-conjugated secondary antibodies and visualized using an enhanced chemiluminescence detection system (GE Healthcare, Piscataway, NJ, USA). The density of the bands was quantified using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Quantitative data are expressed as the mean±standard error. The statistical analysis was performed using SPSS version 12.0 software (SPSS Inc., Chicago, IL, USA) and sigma plot software. The data were analyzed using Student’s t test or one-way analysis of variance, and multiple comparisons among the experimental groups were investigated with the Bonferroni correction test. P values <0.05 were considered to be statistically significant. All the analyses were performed in triplicate.
RESULTS
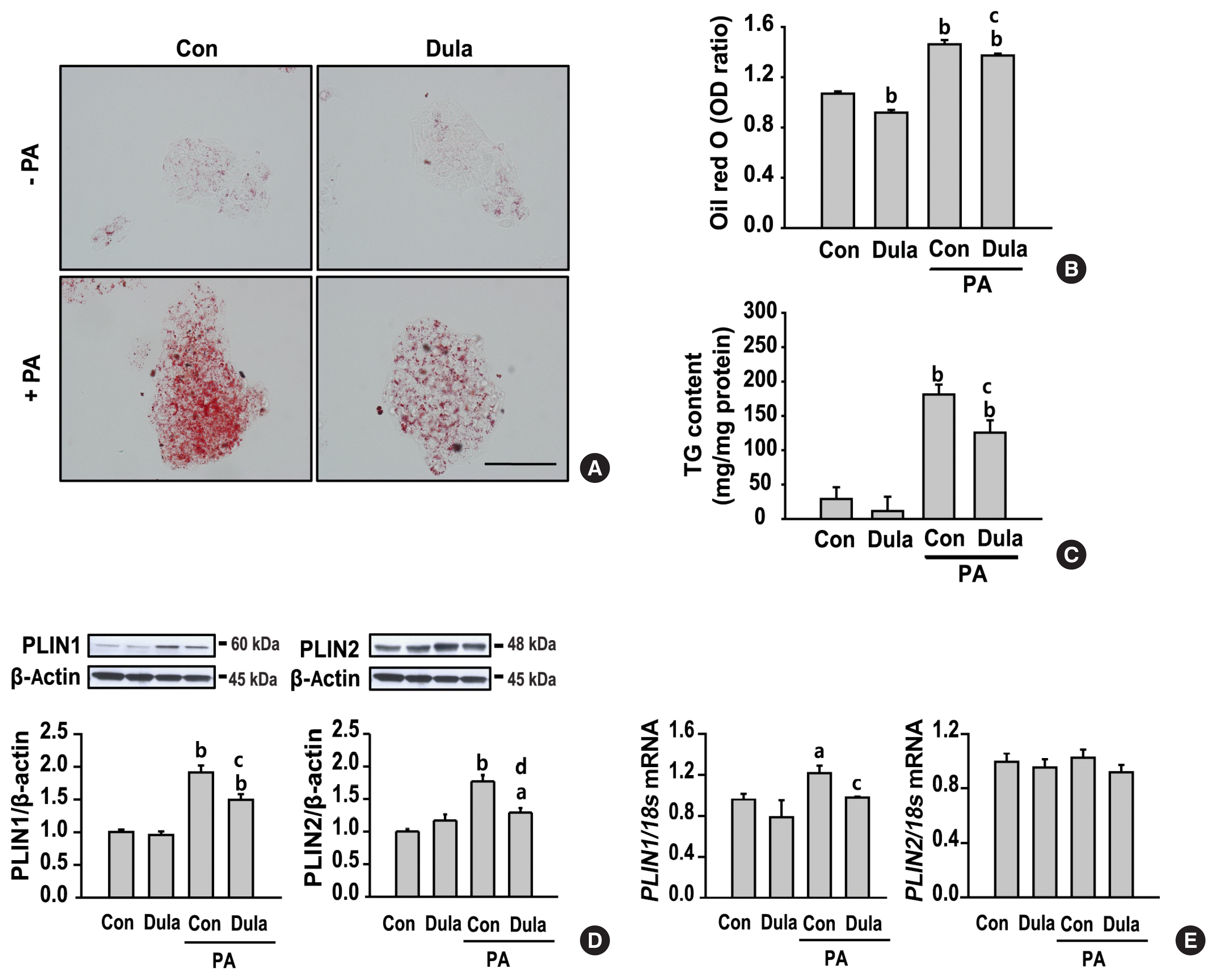
Dulaglutide reduces palmitic acid-induced lipid accumulation in HepG2
To examine the protective effect of dulaglutide on hepatic steatosis, HepG2 cells were treated with a saturated fatty acid, PA. PA significantly increased intracellular lipid accumulation (Fig. 1A–C), which was revealed by Oil red O staining and measurement of TG content. In addition, PA dramatically increased the expression of lipid droplet binding proteins, PLIN1 and PLIN2, which help prevent lipolysis, compared with that in control cells (Fig. 1D, E). However, the mRNA expression of PLIN2 was not significantly affected by PA. In contrast, dulaglutide inhibited TG accumulation and the mRNA and protein expression of PLIN1 and protein expression of PLIN2 in cells pretreated with PA. No significant difference in the expression of PLIN2 mRNA was observed between the cells treated with PA and PA+dulaglutide. These results suggest that dulaglutide has a protective effect against PA-induced hepatic steatosis.
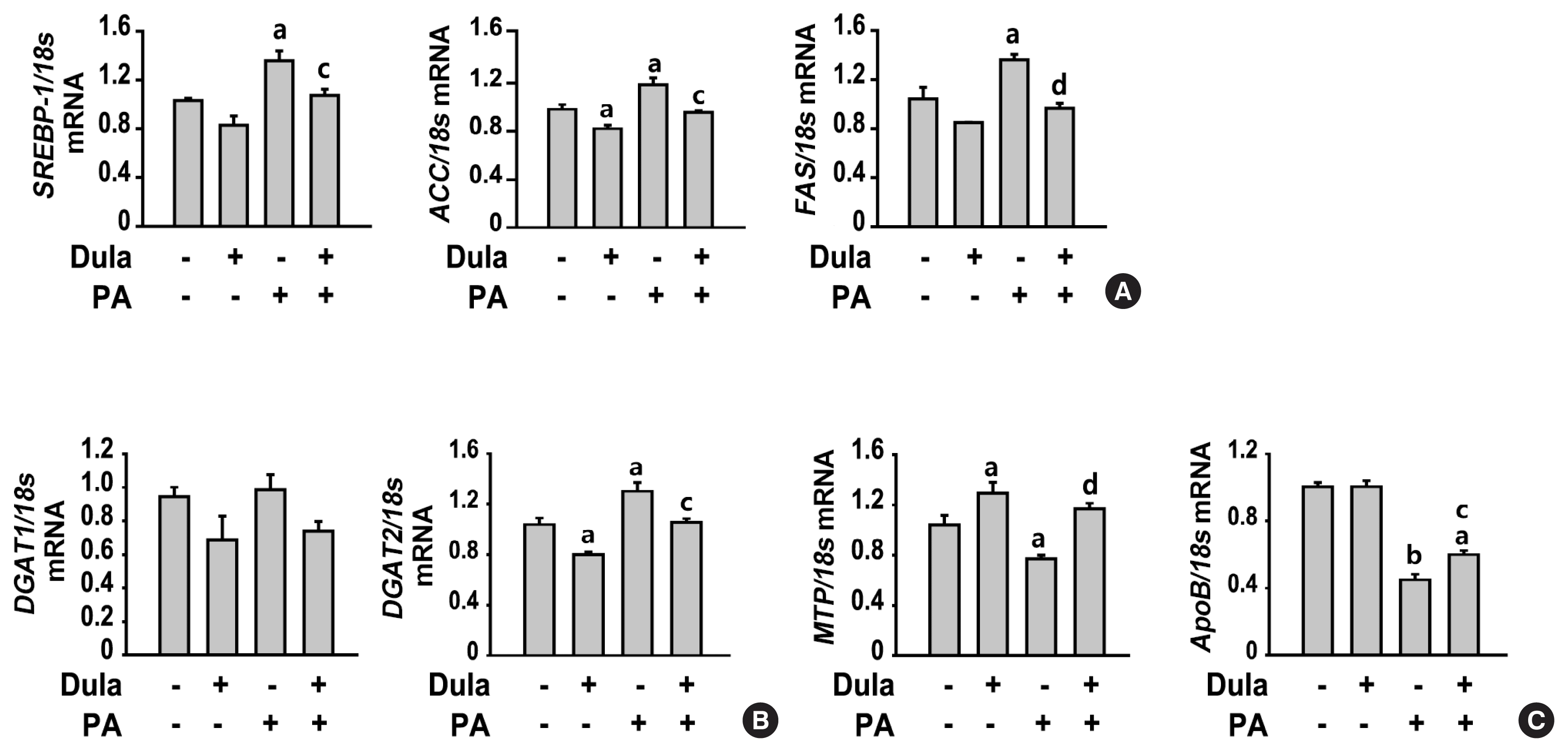
Dulaglutide decreases lipogenesis and TG synthesis and increases TG secretion, lipolysis, and fatty acid oxidation
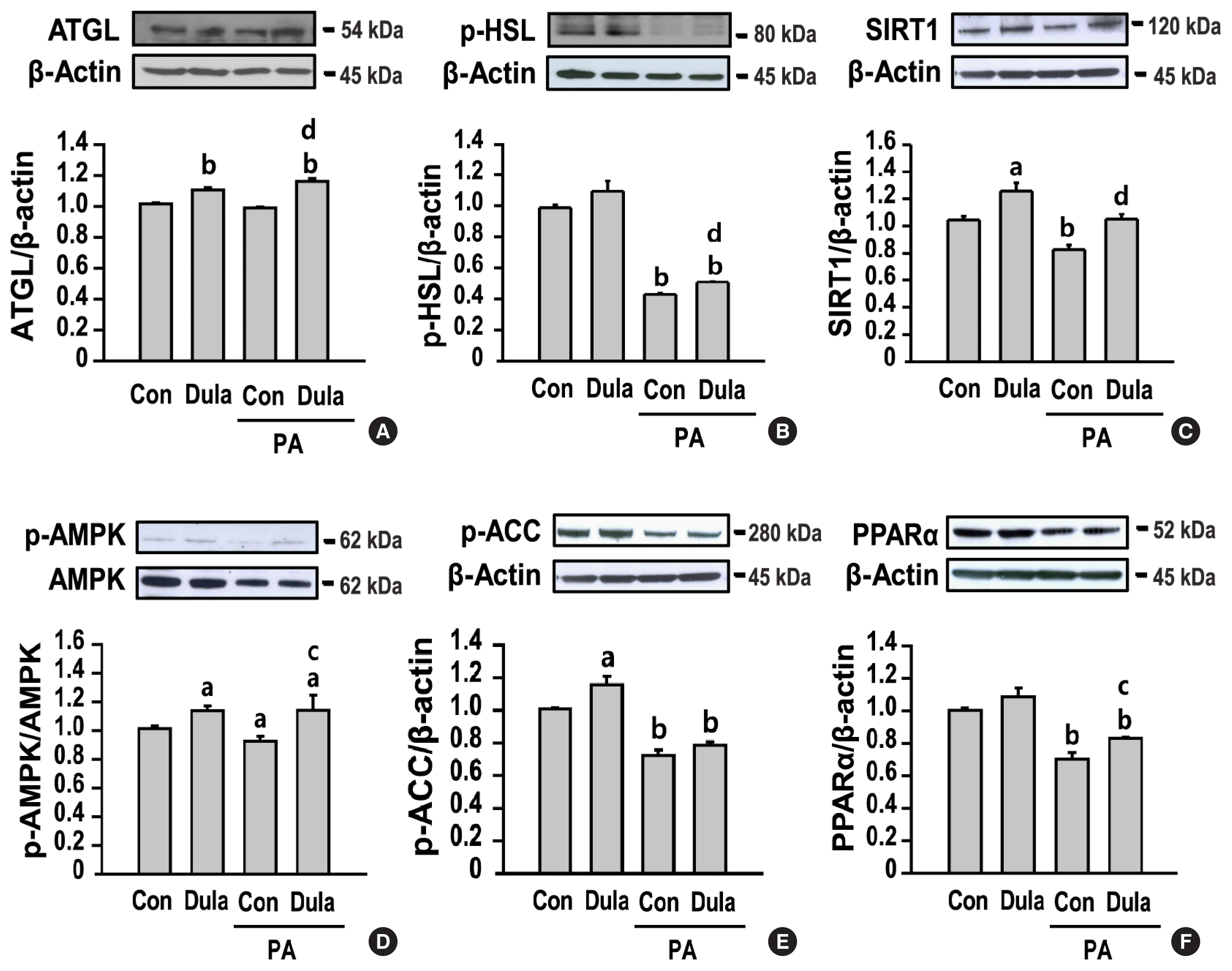
The liver is an important site for lipid metabolism, and hepatic lipid homeostasis is maintained through the balance of synthesis, degradation, and secretion of lipid [19]. We confirmed the change in the mRNA expression of lipogenic genes, including sterol regulatory-binding protein 1 (SREBP-1), ACC, and fatty acid synthase (FAS), the TG synthesis enzyme genes, including diacylglycerol acyltransferase (DGAT) 1 and 2, and key factor genes associated with TG secretion, including microsomal triglyceride transfer protein (MTP) and apolipoprotein B (ApoB). PA increased the expression of SREBP-1, ACC, FAS, and DGAT2 and decreased the expression of MTP and ApoB compared with that in control cells, but no significant change was observed in the expression of DGAT1 (Fig. 2). In contrast, treatment with dulaglutide significantly decreased the expression of SREBP-1, ACC, FAS, and DGAT2 and increased the expression of MTP and ApoB compared with that in the PA-treated cells. We examined whether dulaglutide regulates lipolysis and fatty acid oxidation in PA-treated cells. PA reduced the expression of p-HSL, but not ATGL, which are key enzymes involved in intracellular degradation of TG, and the expression of fatty acid oxidation-associated factors such as SIRT1, p-AMPK, p-ACC, and PPARα, compared with that in control cells (Fig. 3). Meanwhile, treatment with dulaglutide significantly increased the expression of ATGL, p-HSL, SIRT1, p-AMPK, and PPARα proteins compared with that in the PA-treated cells. Therefore, these results suggest that the beneficial effect of dulaglutide against PA-induced hepatic steatosis might be mediated by increasing lipid catabolism and decreasing lipid anabolism.
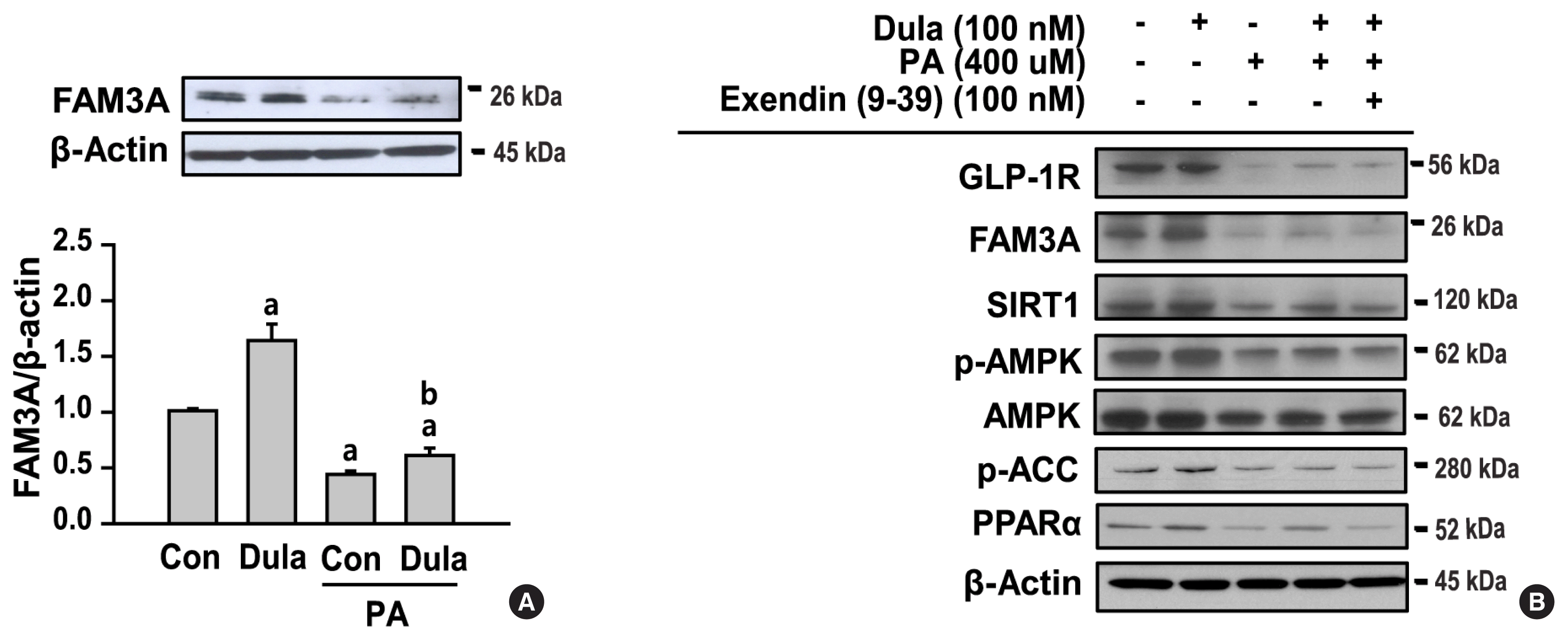
Dulaglutide increases FAM3A expression and fatty acid oxidation through a GLP-1R dependent pathway
An increase in the expression of FAM3A in the liver is associated with improvement in hyperglycemia, insulin resistance, and fatty liver [15]. As shown in Fig. 4A, the expression of FAM3A was decreased in cells treated with PA compared with that in control cells, and increased in cells treated with PA+dulaglutide compared to the cells treated with PA alone. To investigate whether the regulation of FAM3A expression by dulaglutide is mediated by the GLP-1R, HepG2 cells pre-treated with PA were treated with dulaglutide in the absence or presence of exendin (9–39), a GLP-1R antagonist. The protein expression of GLP-1R, FAM3A, SIRT1, p-AMPK, p-ACC, and PPARα was decreased in cells treated with PA compared to the untreated control, and was increased in cell treated with PA+dulaglutide compared to the cells treated with PA alone (Fig. 4B). However, in the presence of exendin (9–39), the increase in GLP-1R, FAM3A, SIRT1, p-AMPK, p-ACC, and PPARα expression in cells treated with PA+dulaglutide was attenuated. Therefore, these results suggest that dulaglutide regulates the expression of FAM3A and fatty acid oxidation-associated factors via a GLP-1R dependent pathway.
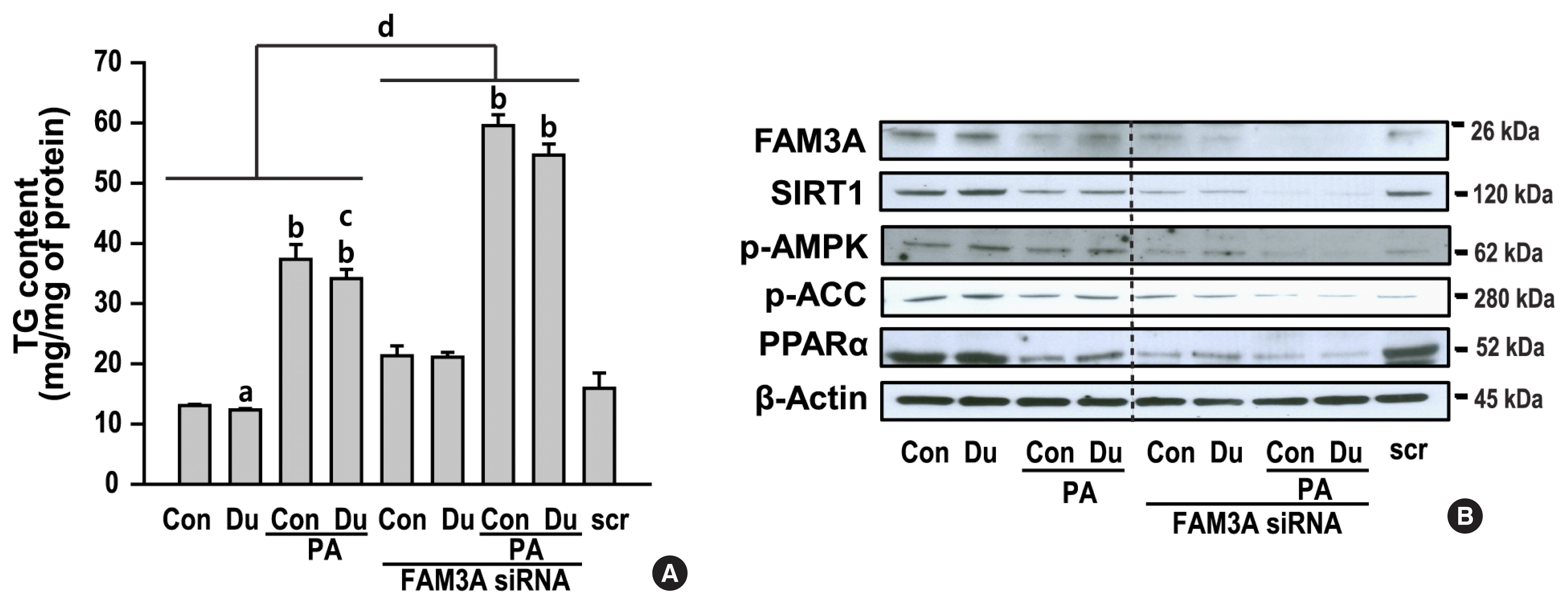
Dulaglutide inhibits TG accumulation and increases fatty acid oxidation by activating FAM3A
To clarity the role of FAM3A in dulaglutide-regulated lipid metabolism in PA-induced hepatic steatosis, HepG2 cells were transfected with the FAM3A siRNA. Inhibition of FAM3A by the siRNA resulted in an increase in TG accumulation; however, treatment with dulaglutide did not decrease TG accumulation in PA-treated cells transfected with FAM3A siRNA (Fig. 5A, Supplemental Fig. S1). In addition, the expression of SIRT1, p-AMPK, p-ACC, and PPARα was decreased in cells transfected with FAM3A siRNA, but dulaglutide treatment did not increase the expression of these proteins in cells transfected with FAM3A siRNA (Fig. 5B). These results suggest that inhibition of TG accumulation by dulaglutide in PA-treated cells is mediated by FAM3A, and that FAM3A expression increased by dulaglutide is involved in the regulation of the expression of SIRT1/AMPK signaling, which is associated with fatty acid oxidation.
DISCUSSION
In this study, we demonstrated for the first time that dulaglutide improves PA-induced hepatic steatosis by increasing TG secretion, lipolysis, and fatty acid oxidation and decreasing lipogenesis and TG synthesis. We also demonstrated that FAM3A activated by dulaglutide exerts a beneficial effect on hepatic lipid metabolism by activating the SIRT1/AMPK signal pathway that stimulates fatty acid oxidation.
In the liver, excessive TG deposition is associated with insulin resistance and NASH, and fatty liver is induced by disrupted equilibrium between several pathways involved in TG synthesis and clearance. The two DGAT enzymes, DGAT1 and DGAT2, are involved in TG synthesis from a variety of fatty acyl-CoA and diglycerides (DG) substrates [20]. DGAT2 synthesizes TG using nascent DG and de novo synthesized fatty acids as substrates, while DGAT1 re-esterifies DG derived from lipolysis to TG [21,22]. DGAT2 has a higher affinity for its substrates than DGAT1, and overexpression of DGAT2 markedly induces increased TG accumulation [20]. We observed that dulaglutide significantly reduced the expression of lipogenic genes and DGAT2, but not DGAT1, in PA-treated HepG2 cells. Furthermore, dulaglutide decreased the expression of lipid droplet binding proteins, PLIN1 and ADFP, also known as PLIN2, and increased the expression of lipases such as ATGL and HSL. The perilipin proteins protect TG from lipolysis by blocking the access of lipases to the lipid droplet [23]. Previous studies have reported that perilipin family members are associated with NAFLD and obesity risk in humans [24–26]. In mice, PLIN2 deletion prevents obesity, insulin resistance, adipose inflammation, and hepatic steatosis [27–29]. These findings suggest that the protective effects of dulaglutide against PA-induced hepatic steatosis are mediated by inhibition of the TG anabolic pathway and by activation of the TG catabolic pathway.
Dulaglutide, a GLP-1RA, is an antidiabetic drug used for the management of type 2 diabetes. Dulaglutide improved NAFLD in patients with type 2 diabetes [30,31]. GLP-1 and GLP-1-based drugs exert therapeutic effects against NAFLD by regulating weight loss, decreasing HbA1c levels, and improving lipotoxicity or insulin sensitivity [32]. However, the mechanism by which dulaglutide reduces hepatic lipid deposition is unknown. Dulaglutide is 90% homologous with native GLP-1, while exendin-4, an another GLP-1RA, has 53% sequence homology with that of GLP-1 [33]. In our previous study, we showed that exendin-4 ameliorates steatohepatitis via the SIRT1/AMPK pathway, which stimulates fatty acid oxidation and is a known potential therapeutic target for the treatment of NAFLD [34]. In this study, we showed that dulaglutide activates the SIRT1/AMPK pathway in PA-treated HepG2 cells and that the expression of the SIRT1/AMPK pathway in cells treated with dulaglutide is regulated by FAM3A. Wang et al. [15] reported that FAM3A increases mitochondrial adenosine triphosphate (ATP) production and secretion, which activates phosphoinositide 3-kinase/AKT in a calcium/calmodulin-dependent manner, and that these pathway increased by FAM3A represses gluconeogenesis and lipogenesis. AMPK is phosphorylated and activated by two upstream kinases: liver kinase B1 (LKB1), which is regulated by the AMP/ATP ratio, and CaMKKβ, which is regulated by calcium levels. We showed that dulaglutide increases the expression of CaMKKβ compared with that in the PA-treated cells (Supplemental Fig. S2). Therefore, these results suggest that FAM3A increased by dulaglutide could activate AMPK signaling via increasing calcium levels.
FAM3A is a cytokine that is ubiquitously expressed in the tissues of human and rodents [14]. FAM3A controls the expression of pancreas/duodenum homeobox protein 1 (PDX1) and insulin and insulin secretion in the pancreatic beta cells under physiologica l and diabetic conditions [35]. Zhou et al. [36] reported that the expression of FAM3A in human hepatocytes is increased by the PPARγ agonist, but not by PPARα and PPARδ agonists. In this study, we confirmed that FAM3A regulates the expression of PPARα, which is an AMPK downstream target and is involved in fatty acid oxidation, by showing that FAM3A inhibition by siRNA reduces the expression of PPARα. Furthermore, we also showed that the increase in fatty acid oxidation and the decrease in TG content induced by dulaglutide are mediated by increasing FAM3A expression. However, FAM3A expression was increased by the GLP-1RA, dulaglutide, and is reversed by the GLP-1R antagonist, exendin (9–39). These results suggest that the protective effect of dulaglutide on fatty liver is mediated, at least partially, through FAM3A expression, which is regulated by a GLP-1R dependent pathway.
In conclusion, this study showed that dulaglutide ameliorated hepatic steatosis by regulating lipid metabolism. We also found that a reduction in hepatic lipid accumulation by dulaglutide was mediated by FAM3A, a regulator of fatty acid oxidation, in a GLP-1R-dependent manner. Thus, FAM3A could be a potential therapeutic approach for treating NAFLD. However, the role of FAM3A in lipogenesis, TG secretion, TG synthesis, and lipolysis requires further elucidation, and additional animal experiments are needed to further clarify that dulaglutide ameliorates hepatic steatosis by activating the FAM3A signaling pathway.
 XML Download
XML Download