

 PDF
PDF Citation
Citation Print
Print



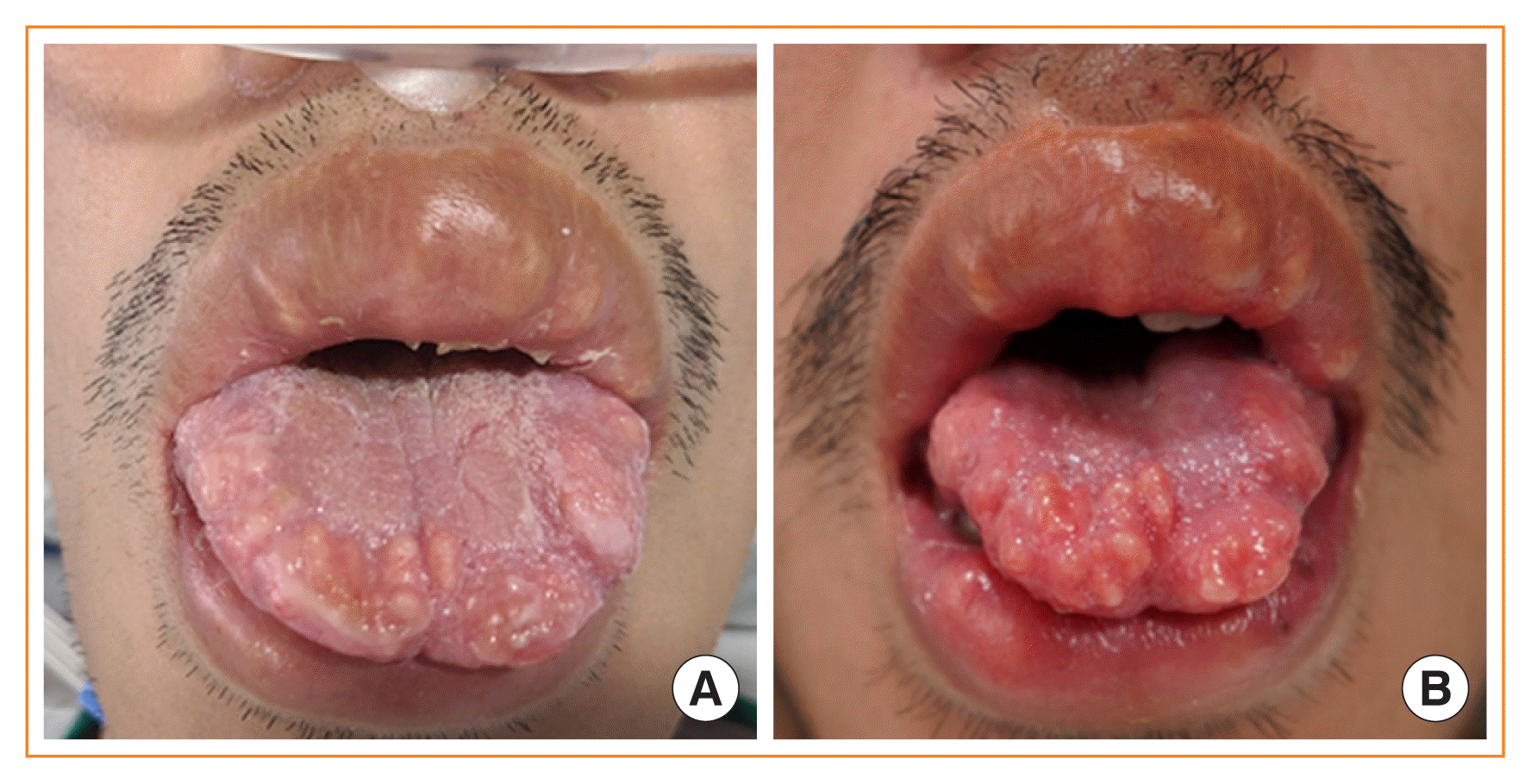
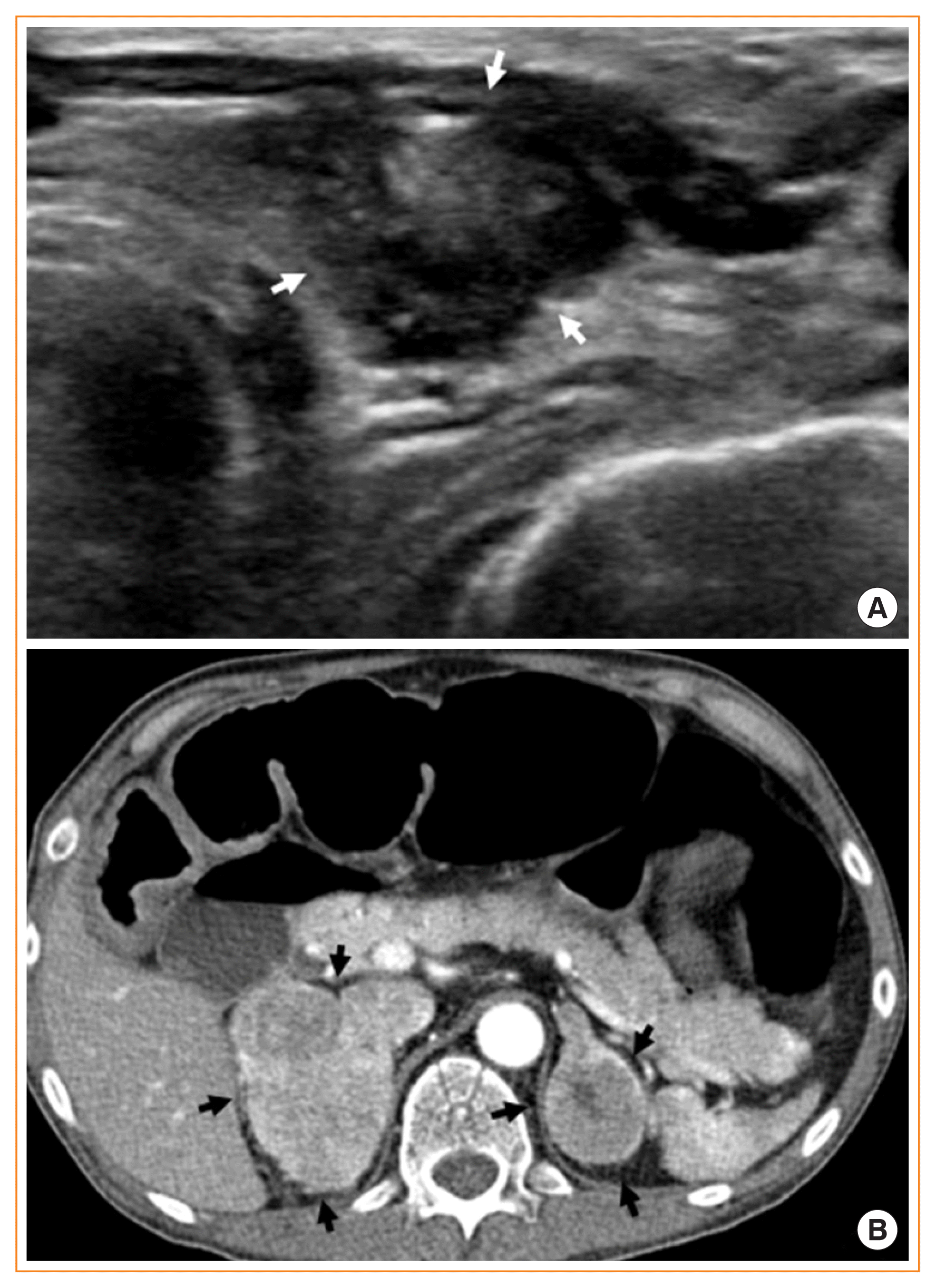
A 34-year-old man presented with poor oral intake and abdominal pain with bilateral adrenal masses on abdominal computed tomography (CT). He had marfanoid features and mild intellectual disability. During the initial physical examination, lingual mucosal neuromas were identified, although they were obscured initially by a cracked and dry tongue, which are signs of dehydration (Fig. 1A); the neuromas with thickened lips were more clearly visible on a photograph taken 6 days after the bilateral adrenalectomy (Fig. 1B). Multiple endocrine neoplasia type 2B (MEN2B) was suspected. He had severe hypertension and hypokalemia of 2.4 mmol/L, a transtubular potassium gradient of 9, and high plasma renin activity, which was uncontrolled despite alpha-blocker and spironolactone treatment. The blood pressure was managed using an additional intravenous calcium channel blocker and beta-blocker infusion therapy, but it was very labile. The accompanying intestinal pseudo-obstruction worsened despite aggressive medical treatments, while the acalculous cholecystitis was managed via percutaneous transhepatic gallbladder drainage (PTGBD). Ectopic adrenocorticotropic hormone (ACTH) syndrome (EAS), a life-threatening endocrine emergency, was suspected based on a serum cortisol level >75 μg/dL, ACTH of 868 pg/mL (reference, 10 to 90), serum cortisol >75 μg/dL after 1 mg overnight dexamethasone suppression, and the results of a corticotropin-releasing hormone stimulation test. A neck ultrasound showed four thyroid nodules, including a 1.8 cm-sized nodule in the left thyroid gland (Fig. 2A), with cervical lymph nodes suspicious for metastasis. Cells aspirated from the nodule tested positive for calcitonin on immunocytochemical staining. Bilateral pheochromocytomas were suggested by abdominal CT (Fig. 2B) and 18F-fluorodeoxyglucose positron emission tomography with CT findings, as well as elevated levels of plasma metanephrine (>20 nmol/L; reference, ≤0.5), plasma normetanephrine (>45 nmol/L; reference, ≤0.9), 24-hour urine vanillylmandelic acid (196 mg/day; reference, ≤8), and 24-hour urine metanephrine (49 mg/day; reference, ≤0.8). Although either a pheochromocytoma or medullary thyroid carcinoma (MTC) are possible causes of EAS in MEN2B [1,2], bilateral adrenalectomy following medical control of intense hypercortisolism and preoperative alpha-blockade was considered first as a definitive treatment for bilateral pheochromocytoma; this can also serve as a rescue treatment in cases of EAS caused by MTC [3]. An etomidate infusion was started in the intensive care unit to inhibit steroidogenesis; 6 days later, bilateral adrenalectomy was performed successfully. After surgery, bowel distension was relieved, the blood pressure and biochemical parameters returned to normal, and the PTGBD could be removed. After obtaining informed consent, RET proto-oncogene analysis confirmed an M918T mutation. Following discharge from the hospital, the patient has been receiving hydrocortisone replacement therapy, and his performance status is improving. Vandetanib was initiated to treat MTC with liver metastasis in which calcitonin levels exceed 8,000 pg/mL and carcinoembryonic antigen levels are elevated (299 ng/mL; reference, 0 to 5). The patient’s calcitonin levels had decreased to 1,322 pg/mL at 1 month after the initiation of treatment. To successfully treat this rare, life-threatening endocrine emergency, a careful physical examination and timely multidisciplinary planning among several departments are essential.
This image of interest was approved by the Institutional Review Board (JNUH IRB approval no. 2021-09-031). Written informed consent was obtained from the patient and a family member to publish this image of interest.
 XML Download
XML Download