

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The development of atherosclerosis involves dyslipidemia, characterized by elevated low-density lipoprotein cholesterol (LDL-C), triglycerides [1], reduced high-density lipoprotein cholesterol (HDL-C), inflammation, and endothelial dysfunction [2]. These metabolic changes are often accompanied by increased fat deposition in the liver and consequent hepatitis, which is characterized by hepatocyte necrosis and fibrosis.
Farnesoid X receptor (FXR) is highly expressed in the liver, intestine, kidneys, and adrenal glands, and is known to be activated by bile acids, including cholic acid and chenodeoxycholic acid [3,4]. Activation of FXR decreases serum triglyceride levels, while FXR deficiency increases plasma levels of triglycerides and total cholesterol levels—both HDL-C, due to reduced clearance, and non-HDL-C—in animal models [5,6].
Based on studies on the effects of FXR on lipid metabolism, further investigations explored the pro-atherogenic effects of FXR. Whereas apolipoprotein E (ApoE)−/− mice are well established as a model of atherosclerosis, resulting from poor lipoprotein clearance and subsequent deposition of lipids in blood vessels [7], FXR−/− mice have not been able to demonstrate an atherosclerotic phenotype despite a proatherogenic profile of serum lipoproteins [8]. In a previous study comparing wild-type mice, ApoE−/− mice, FXR−/− mice, and ApoE−/−FXR−/− mice fed high fat/high cholesterol diets, plasma cholesterol levels showed sequentially increased levels in FXR−/−, ApoE−/−, and ApoE−/− FXR−/− mice, while plasma triglyceride levels were only significantly elevated in FXR−/− and ApoE−/−FXR−/− mice. Atherosclerotic plaques were detectable in ApoE−/− mice and were present at an augmented level in ApoE−/−FXR−/− mice, but not in FXR−/− mice, indicating that the double knockout genotype is necessary to study the role of FXR in atherosclerosis [9].
The activation of FXR by a synthetic agonist reversed diet-induced hypertriglyceridemia and elevations of non-HDL-C, while producing near-complete inhibition of aortic lesion formation in (low density lipoprotein receptor knock out (LDLR−/−) and ApoE−/− mice [10]. Another study also revealed that dual activation of the bile acid nuclear receptor FXR and G protein-coupled bile acid receptor 1 (GPBAR1 or TGR5) protected mice against atherosclerosis and reduced both circulating lipid levels and inflammation, strengthening the evidence for a relationship between FXR and atherosclerosis [11]. In addition, FXR downregulation has been implicated in the development of nonalcoholic fatty liver disease (NAFLD) [12], and FXR agonists were shown to improve hepatic fibrosis and inflammation in in vivo studies [13].
Peroxisome proliferator-activated receptors (PPARs) are well known modifiers of insulin sensitivity, lipid metabolism, and inflammation. PPARα is known to exert effects on multiple levels of fatty acid metabolism, and PPARγ is known to regulate adipose genes to promote lipolysis and clear triglyceride-rich lipoproteins, suggesting that they play favorable roles in various settings of altered lipid metabolism [14].
Thus, the aim of this study was to investigate the mechanisms of FXR-dependent atherosclerosis and NAFLD in detail, as well as the roles of PPARα and PPARγ agonists in this setting.
METHODS
Animals and diets
Both ApoE−/− and ApoE−/−FXR−/− mice (C57BL/6J) were provided by Professor Young Joo Park (Department of Internal Medicine, Seoul National University College of Medicine, Seoul, Korea). To generate ApoE−/−FXR−/− mice, homozygous knockout mice for ApoE or FXR were first generated by crossing heterozygous knock out mice for each gene. After confirming the selection by genotyping, the generated ApoE−/− mice and FXR−/− mice were subsequently crossed to generate ApoE−/−FXR−/− mice. The mice were fed with a Western diet (WD; 17% protein, 40% fat, 43% carbohydrate; Research Diets Inc., New Brunswick, NJ, USA). Pioglitazone and fenofibrate were administered via oral gavage at doses of 20 and 100 mg/kg for pioglitazone [15-18] and fenofibrate [19-21], respectively, to provide a moderately high dose based on safely tolerated doses in other mouse models in the previous literature for each drug.
At 8 weeks of age, the ApoE−/− mice (n=7) were kept on the WD for 12 weeks. The ApoE−/−FXR−/− mice (n=25) were divided into three groups and were kept on the WD alone (n=9), treated with pioglitazone (n=8), or treated with fenofibrate (n=8) for 12 weeks. Mice were weighed once every two weeks, and food consumption was monitored throughout the study. All animals were maintained at 23°C±2°C and 60%±10% humidity on a 12-hour light/dark cycle, and food and water were provided ad libitum. At the end of the study, mice were fasted for 8 hours, anesthetized, and sacrificed.
All procedures were conducted in accordance with the Guide for Standard Operation Procedures and with approval of the Institutional Animal Care and Use Committee (IACUC) in the Clinical Research Institute of Seoul National University Bundang Hospital (BA1407-156/030-03).
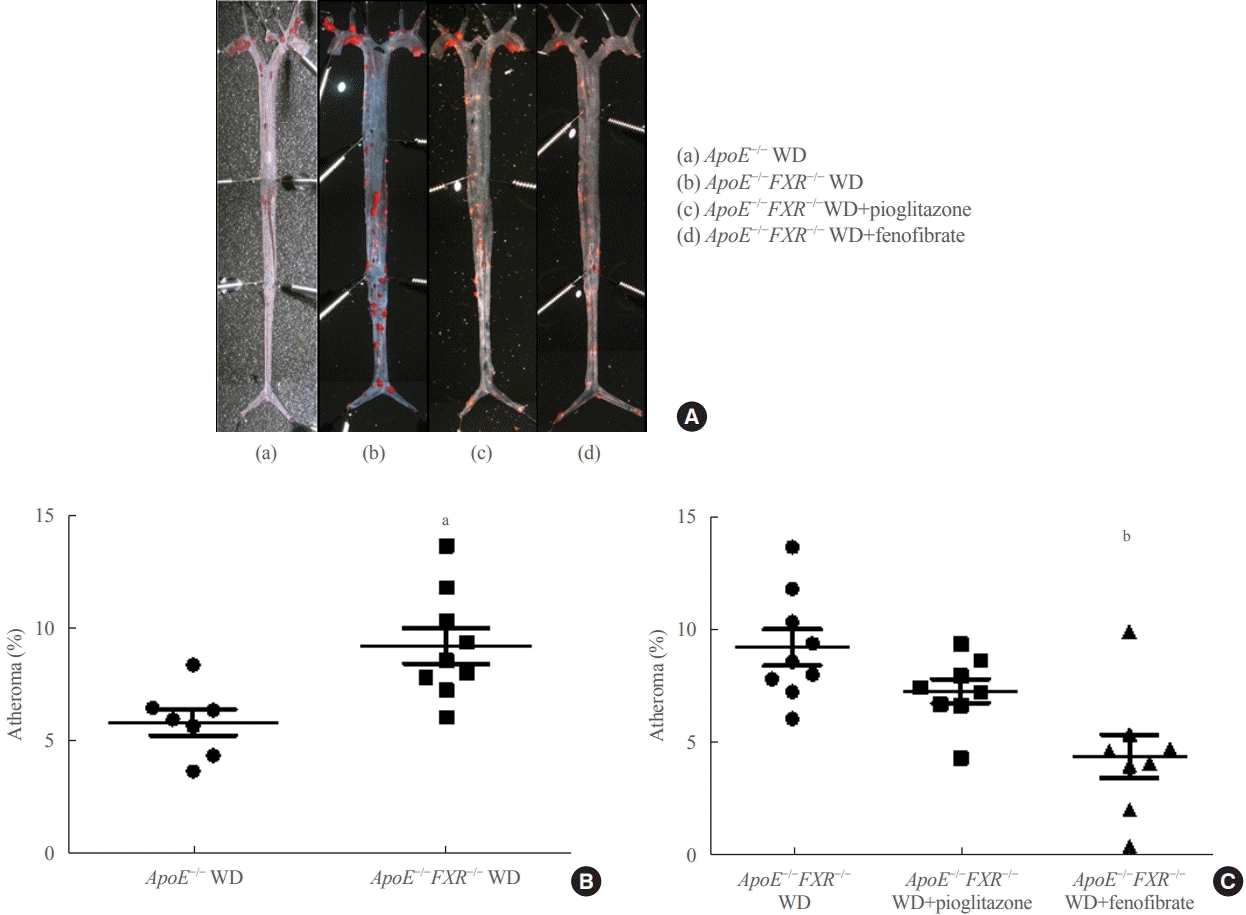
En face analysis
En face lipid accumulation was determined by removing aortas from the mice from the ileal bifurcation to the origin at the heart. The whole aorta was fixed in 4% phosphate buffered saline for 48 hours, cut longitudinally, pinned on a plate, and stained with Oil Red O to identify atherosclerotic lesions. Images were taken using a light microscope (Carl Zeiss, Oberkochen, Germany) and analyzed. Atherosclerotic lesions of the aorta were presented as the percentage of the sum of the Oil Red Opositive area to the total aortic surface area.
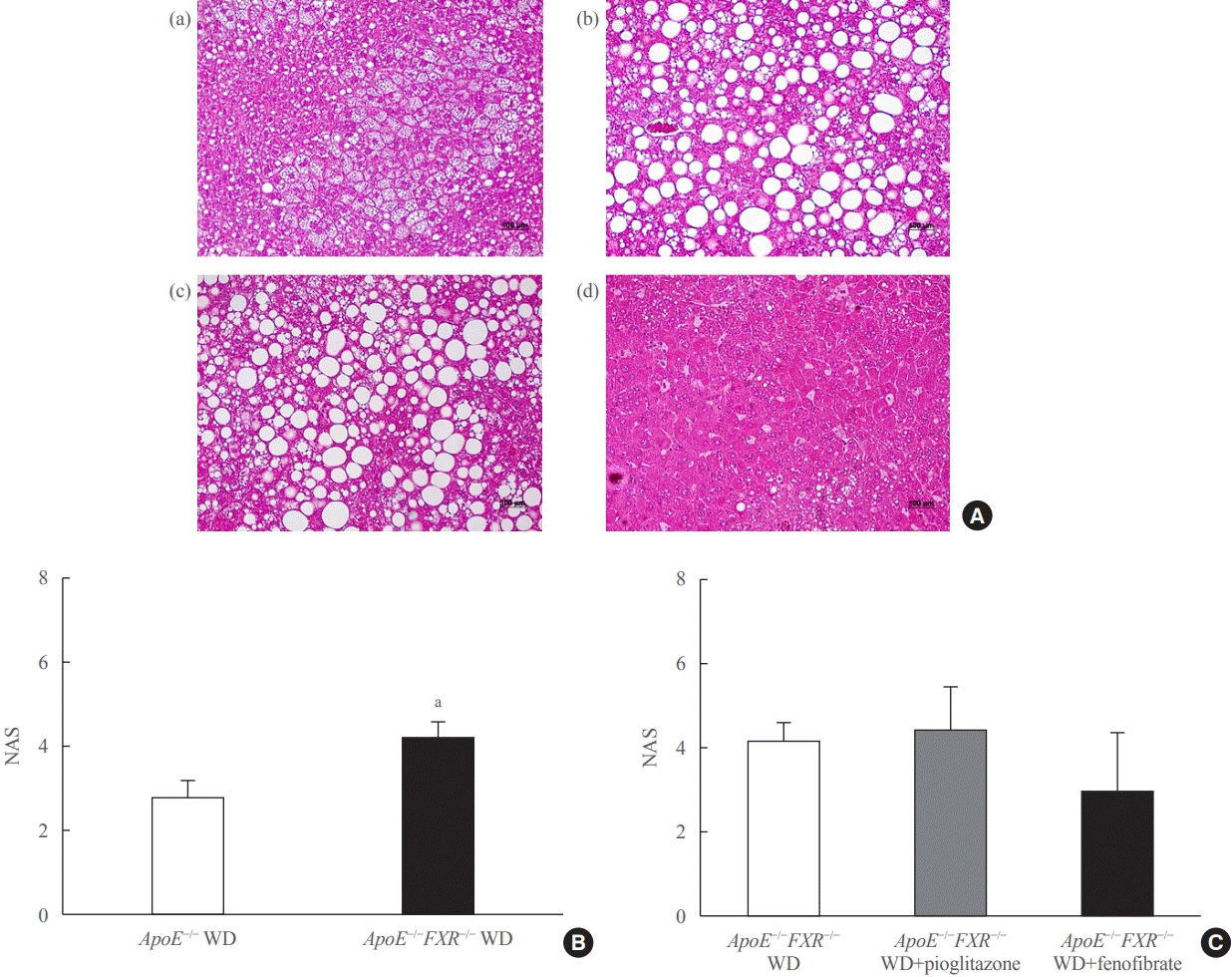
Liver histology
Liver tissue was fixed in 4% formaldehyde, dehydrated, embedded in paraffin, and sectioned (4 μm). Sections were stained with hematoxylin and eosin and Masson trichrome. The NAFLD activity score (NAS) and fibrosis score were calculated by an experienced pathologist. The NAS is defined as the sum of the scores for steatosis (0 to 3), lobular inflammation (0 to 3), and ballooning (0 to 2). The NAS ranges from 0 to 8 and a score equal to or greater than 5 is correlated with a diagnosis of nonalcoholic steatohepatitis. Fibrosis, which is thought to be a result of the disease rather than a reversible factor that indicates activity, was separately staged from 0 to 4 (0, none; 1, perisinusoidal or periportal; 1A, mild, zone 3, perisinusoidal; 1B, moderate, zone 3; perisinusoidal, 1C, portal/periportal; 3, bridging fibrosis; and 4, cirrhosis) [22].
Serum glucose and lipid measurements
Blood was obtained from the inferior vena cava after overnight fasting. Serum was prepared and stored at –80°C. Blood glucose levels were determined by a glucometer (ACCU-CHEK Active, Roche, Mannheim, Germany). Total cholesterol, triglyceride, HDL-C, and LDL-C levels were determined by the Beckman Coulter AU480 automatic biochemistry analysis system (Tokyo, Japan).
Quantitative real-time polymerase chain reaction
Total RNA was isolated from the liver, white adipose tissue, and gastrocnemius muscle using TRizol reagent (Ambion, Foster City, CA, USA), and cDNA was synthesized using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Reverse-transcriptase quantitative polymerase chain reaction (RT-qPCR) was performed using the ABI 7500 Real Time PCR System (Applied Biosystems, Foster City, CA, USA) and amplification was achieved using the SYBR Premix Ex Taq polymerase (Takara, Otsu, Japan). Genespecific primers (Cosmo Genetech, Seoul, Korea) for selected genes were designed using Primer Express software (Applied Biosystems). The selected genes and their primer sequences are included in the supplementary material (Supplemental Table S1). All resulting data were analyzed using ABI 7500 software version 2.0.5 (Applied Biosystems).
RESULTS
Loss of FXR in ApoE-deficient mice increases atherosclerosis, which is reversed by a PPARα agonist
The percentage of atherosclerosis was significantly higher in ApoE−/−FXR−/− mice than in ApoE−/− mice (5.9%±1.5% vs. 9.2%±2.4%, P=0.006) (Fig. 1A, B) even after weight adjustment (P=0.033). The increased atherosclerosis in ApoE−/− FXR−/− mice was reversed by PPARα agonist (fenofibrate) treatment (9.2%±2.4% vs. 4.4%±2.7% in WD controls, P=0.001) but not by PPARγ agonist (pioglitazone) treatment (9.2%±2.4% vs. 7.3%±1.5% in WD controls, P=0.216) (Fig. 1C).
Loss of FXR in ApoE-deficient mice increases serum levels of total cholesterol, triglyceride, and LDL-C levels
ApoE−/−FXR−/− mice had higher serum levels of total cholesterol (1,091±176 mg/dL vs. 691±152 mg/dL, P=0.017), triglycerides (289±52 mg/dL vs. 179±49 mg/dL, P=0.016), and LDL-C (798±135 mg/dL vs. 574±79 mg/dL, P=0.008) than ApoE−/− mice. However, serum HDL-C levels did not differ significantly between the two groups (54±22 mg/dL vs. 47±27 mg/dL, P=0.683).
Treatment with a PPARα agonist decreases serum triglyceride levels in ApoE−/−FXR−/− mice
Treatment with fenofibrate decreased serum triglyceride level in ApoE−/−FXR−/− mice (190±73 mg/dL vs. 289 ±52 mg/dL in WD controls, P=0.028), whereas treatment with pioglitazone did not (310±35 mg/dL vs. 289 ±52 mg/dL in WD controls, P=0.874 in the post hoc analysis). Serum levels of total cholesterol, LDL-C, and HDL-C were unaffected by either treatment. Serum glucose levels were not different regardless of genetic background and treatment (Supplemental Table S2).
Loss of FXR in ApoE-deficient mice aggravates liver steatosis, which is reversed by PPARα treatment
ApoE−/− mice showed predominantly microvesicular steatosis characterized by distended hepatocytes with foamy-appearing cytoplasm due to small lipid vesicles. In ApoE−/−FXR−/− mice, the liver showed predominantly macrovesicular steatosis characterized by distortion of the nucleus due to the large size of the lipid vesicles. ApoE−/−FXR−/− mice also demonstrated mild to moderate perivenular/perisinusoidal or periportal fibrosis (stages 1A to 2), in contrast to the ApoE−/− mice with no fibrosis (Fig. 2A, B, Supplemental Fig. S1A)
Treatment with fenofibrate significantly improved the degree of steatosis, but not the necroinflammatory changes or the NAS, which is a sum of scores assessing steatosis (score 0 to 3), lobular inflammation (0 to 3), and hepatocyte ballooning (0 to 2); thus ranging from 0 to 8. Treatment with pioglitazone improved neither steatosis nor the lobular necroinflammation (Fig. 2A, C, Supplemental Fig. S1B). No significant differences were observed in the degree of fibrosis among the ApoE−/−FXR−/− mice according to the treatment (data not shown).
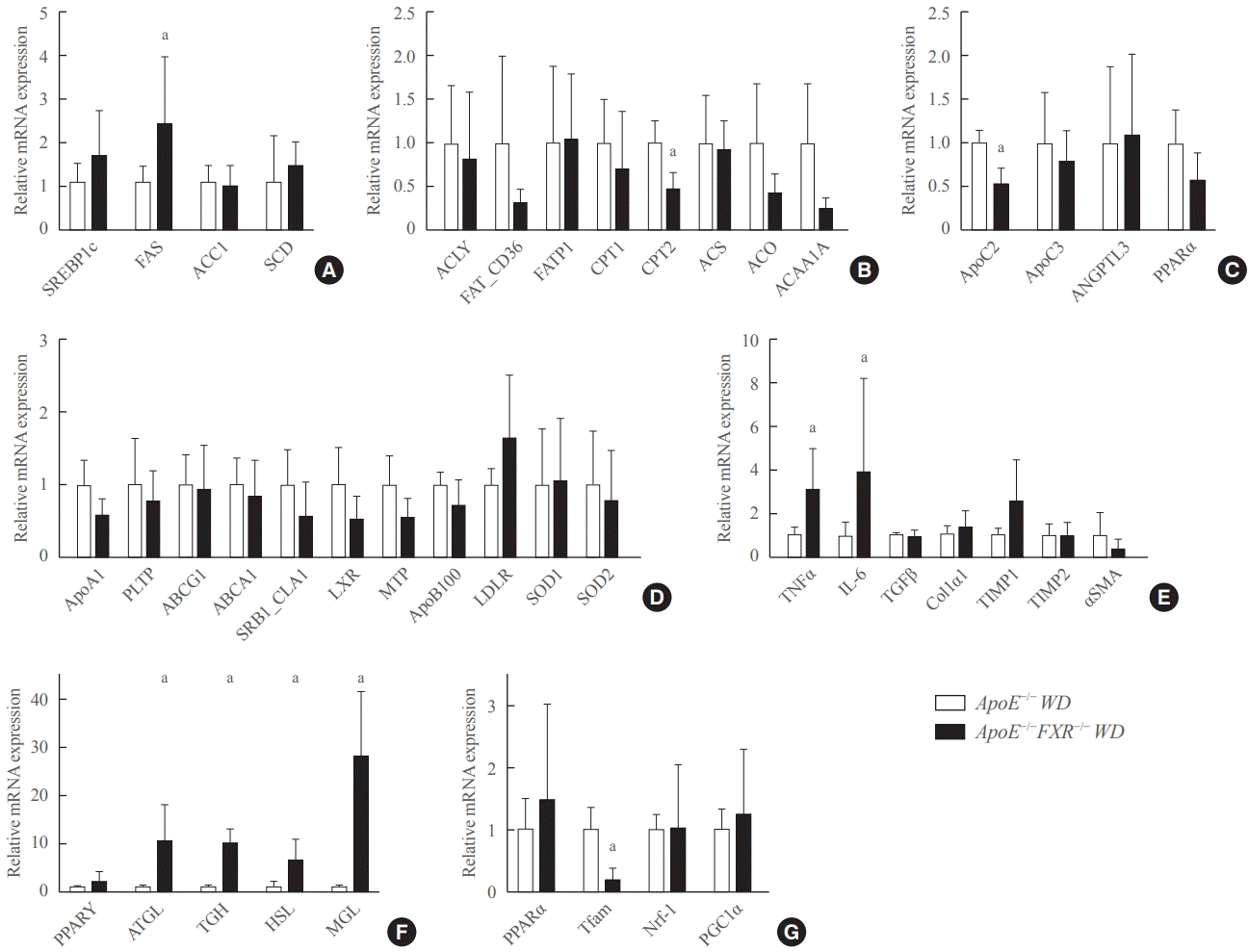
FXR deficiency is associated with altered fatty acid synthesis, triglyceride hydrolysis, and increased inflammatory cytokines in the liver
Among the genes involved in fatty acid metabolism, the expression of fatty acid synthase (FAS) was significantly elevated and that of carnitine palmitoyltranferase 2 (CPT2) was significantly reduced in ApoE−/−FXR−/− mice compared to ApoE−/− mice (Fig. 3A, B). In the analysis of genes involved in triglyceride metabolism, ApoC2, which is known to activate lipoprotein lipase (LPL), was significantly decreased in ApoE−/−FXR−/− mice (Fig. 3C). An analysis of hepatic genes involving very LDL metabolism and LDL metabolism did not reveal any differences between ApoE−/− and ApoE−/−FXR−/− mice (Fig. 3D). The expression of genes for the inflammatory cytokines tumor necrosis factor-α (TNFα) and interleukin-6 (IL-6) was also significantly elevated in ApoE−/−FXR−/− mice (Fig. 3E). However, genes related to hepatic fibrosis were not altered (Fig. 3E).
FXR deficiency increases lipolysis in adipose tissue and decreases mitochondrial activation in the skeletal muscle
The RT-qPCR analysis of genes involved in lipolysis in adipocytes showed markedly increased expression of adipocyte triglyceride lipase (ATGL), triglycerol hydrolase (TGH), hormone sensitive lipase (HSL), and monoglyceride lipase (MGL) (Fig. 3F). In the analysis of skeletal muscle genes, transcription factor A, mitochondrial (Tfam), which is involved in mitochondrial activation, was significantly decreased (Fig. 3G).
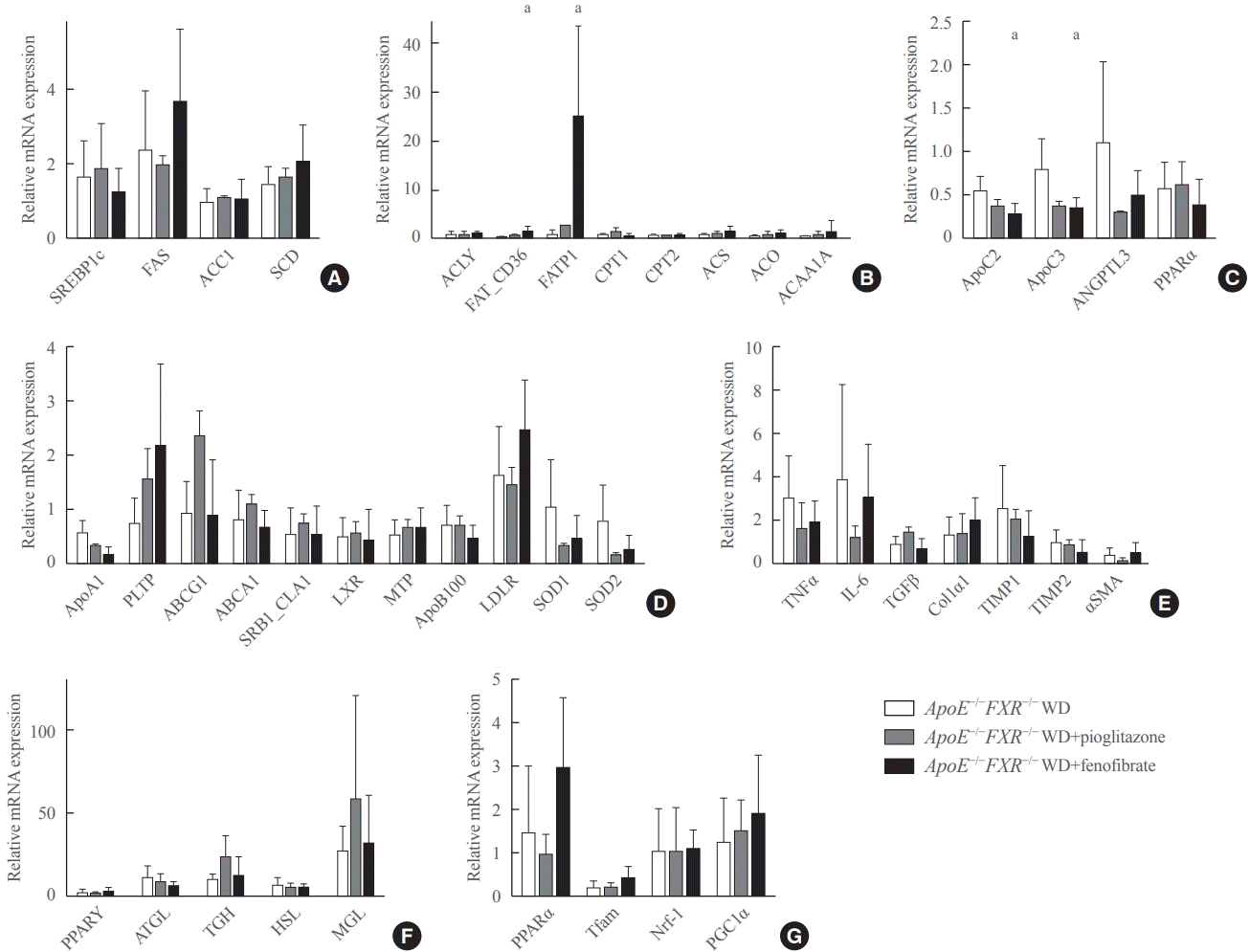
Treatment with a PPARα agonist increases fatty acid uptake and triglyceride hydrolysis in the liver of ApoE−/−FXR−/− mice
Fenofibrate treatment significantly increased the expression of CD36 (fatty acid translocase [FAT]) and fatty acid transport protein 1 (FATP1) in ApoE−/−FXR−/− mice (Fig. 4B), while not altering the genes related to fatty acid synthesis (Fig. 4A). Moreover, an analysis of genes involved in triglyceride hydrolysis showed that fenofibrate decreased levels of both ApoC2 and ApoC3, which are respectively activator and inhibitor genes of LPL, in ApoE−/−FXR−/− mice (Fig. 4C). Fenofibrate treatment did not affect the expression of genes involved in cholesterol metabolism, inflammation, hepatic fibrosis, adipocyte lipolysis, and mitochondrial activation (Fig. 4D-G). Pioglitazone treatment was not associated with improvement of any of the genes related to lipid metabolism (Fig. 4D-G).
DISCUSSION
This study showed that loss of FXR was associated with aggravation of atherosclerosis and hepatic steatosis in ApoE-deficient mice, which could be reversed by a PPARα agonist through induction of triglyceride hydrolysis and β-oxidation of fatty acids.
Treatment of ApoE−/−FXR−/− mice with a PPARα agonist (fenofibrate) showed a significant decrease in atherosclerotic lesions and hepatic fat accumulation, but treatment with a PPARγ agonist (pioglitazone) did not. Neither of the PPAR agonists caused significant changes in the degree of hepatic necroinflammation, ballooning, or fibrosis. Biochemically, reduction of serum triglyceride levels was the only factor that could account for the improvement in atherosclerosis and hepatic steatosis caused by the loss of FXR in fenofibrate-treated mice.
To explain the possible mechanisms for the above metabolic changes, we performed an RT-qPCR analysis of related genes. The examination of genes related to fatty acid synthesis, fatty acid uptake/catabolism, and triglyceride hydrolysis showed higher expression of FAS and lower expression of CPT2 and ApoC2 in ApoE−/−FXR−/− mice compared with ApoE−/− mice.
FAS is a known target gene of FXR, which is downregulated upon activation of FXR in the process of lipolysis [23,24]. The elevated expression of FAS in ApoE−/−FXR−/− mice implies that increased lipogenesis contributes to the elevated levels of triglycerides, in turn promoting atherosclerosis. CPTs are responsible for the mitochondrial uptake of the esterified fatty acids in preparation for β-oxidation. The decrease in CPT2 expression in ApoE−/−FXR−/− mice implies that decreased mitochondrial uptake of fatty acids might contribute to decreased fatty acid oxidation, resulting in a surplus of fatty acids in the liver and circulation. In addition, a significant reduction in the expression of ApoC2, which is an LPL activator gene, suggests the contribution of the loss of FXR to reduced triglyceride clearance.
The examination of genes related to inflammation showed higher expression of TNFα and IL-6 in ApoE−/−FXR−/− mice than in ApoE−/− mice, confirming the suppressive role of FXR on inflammation. The investigations of adipocyte lipolysis showed marked elevations of ATGL, TGH, HSL, and MGL in ApoE−/− FXR−/− mice. Previously, FXR agonists were shown to enhance insulin signaling in differentiated 3T3-L1 adipocytes, whereas FXR−/− mice showed decreased insulin sensitivity [6]. Insulin resistance in adipocytes promotes lipolysis and free fatty acid release in circulation [25]. These free fatty acids act on the liver to disrupt the actions of insulin on glucose while retaining its lipogenic actions, increasing de novo lipogenesis and re-esterification of free fatty acids in the form of triglycerides, which contributes to liver fat accumulation that further impairs insulin sensitivity and mitochondrial function. Accordingly, the analysis of mitochondrial activation genes measured in skeletal muscle tissue showed a significant decrease in Tfam in ApoE−/− FXR−/− mice, implying interdependent actions of insulin resistance, lipolysis, liver steatosis, and mitochondrial dysfunction resulting from the loss of FXR in ApoE−/− mice.
The above genes were again analyzed and compared after treatment with a PPAR agonist, and several changes were noted after PPARα agonist treatment. In the analysis of genes related to fatty acid synthesis and uptake/catabolism, increased expression of CD36 and FATP1, which are regulator genes for the cellular uptake of circulating fatty acids, was noted. In relation to TG clearance, ApoC2 and ApoC3, which are genes for positive and negative regulators of LPL, respectively, both showed lower levels of expression. The downregulation of ApoC2 and ApoC3, with a net effect of LPL activation, has been documented in previous studies. ApoC3 is a major constituent of the triglyceride-rich remnant lipoprotein, which impedes lipolysis by inhibiting LPL action. It can be presumed that the increased expression of ApoC3 with a reduction of the ApoC3/ApoC2 ratio may produce a net stimulatory effect on triglyceride hydrolysis [26-29]. The expression of genes related to adipocyte lipolysis, mitochondrial activation, and inflammation was not affected by fenofibrate treatment; this finding is compatible with the liver histology, which showed no improvements in lobular necroinflammation and NAS.
In short, decreased fatty acid oxidation and increased free fatty acid flux into the liver from adipocyte lipolysis, in addition to increased inflammation in association with insulin resistance, account for the metabolic changes caused by FXR loss. Improvements of these metabolic changes by fenofibrate treatment can be explained by the increased cellular uptake of fatty acids for catabolism in liver and LPL-mediated lipolysis by modulation of its regulators.
Previous experimental studies on atherosclerosis showed that FXR activation by agonists induced vascular smooth muscle cell apoptosis [30] and upregulated the expression of endothelial nitric oxide synthase in vitro [31]. In vivo studies using FXR agonists proved to reduce inflammatory cytokines in aortas of ApoE−/−FXR−/− mice and to reduce atherosclerotic lesions in LDLR−/− and ApoE−/− mice [10,32,33]. Dual activation of FXR with G-protein coupled receptor TGR5 also attenuated atherosclerosis both in LDLR−/− and ApoE−/− mice [11]. In addition, studies on NAFLD showed that FXR activation by natural and synthetic agonists improved hepatic steatohepatitis by reducing fatty acid synthesis via increasing the expressions of SHP, reducing the expression of sterol regulatory element-binding protein 1c (SREBP1c) [23], and increasing cholesterol efflux [34]. FXR agonist also produced a significant improvement of hepatic inflammation in mice [35] and liver fibrosis in humans [13].
Despite these promising results on the effect of FXR agonism in reducing cardiovascular morbidity and mortality, several factors preclude the development of FXR agonists as therapeutic agents. A clinical trial of obeticholic acid in NAFLD showed more episodes of pruritis, reversible heart failure, osteopenia, lower HDL-C levels, higher LDL-C levels, and more severe fatigue compared with placebo group despite improvement of liver histology through FXR agonism [36]. FXR agonists have also been found to cause HDL-C reduction in multiple in vitro studies, constituting a major obstacle to their development as metabolic enhancers [37-39].
In contrast, fenofibrate, a well-known PPARα agonist, has long been used in clinical settings for the treatment of patients with metabolic derangements focusing on triglyceride metabolism, and it has a predictable safety profile. Additionally, incubation of human hepatoma HepG2 cells with natural and synthetic FXR agonists both resulted in a significant induction of PPARα mRNA levels, suggesting crosstalk between FXR and PPARα [40]. These findings imply that fenofibrate may function as a potent alternative to FXR agonists as a therapeutic agent in patients with metabolic derangements related to FXR dysfunction, with lower risks of developing undesirable effects.
In this study, the improvement of serum triglyceride levels by fenofibrate, apart from persistent LDL-C levels, was able to reverse atherosclerosis and hepatic steatosis, although not to the degree of improving NAS. These changes were accompanied by increased fatty acid catabolism and LPL activation, reversing the changes brought by increased lipogenesis and decreased LPL activation in FXR deficiency. Although the results did not show direct crosstalk between FXR and PPARα, our study demonstrated that PPARα agonists can restore some of the metabolic derangements caused by FXR by working on common metabolic pathways to reduce plasma triglyceride levels, thereby improving atherosclerosis and NAFLD.
This study has limitations. First, our investigation focused mainly on triglyceride and free fatty acid metabolism, linking adipocyte lipolysis to the increased lipid accumulation in liver. A more thorough evaluation of fatty acid metabolism, including studies of free fatty acid trafficking with measurements of hepatic uptake of circulating free fatty acids, may better illustrate the links between liver and adipose tissues. Second, the development of atherosclerosis results from altered lipid metabolism in combination with insulin resistance, increased inflammatory response, and endothelial dysfunction. However, changes in inflammation and endothelial dysfunction in blood vessels were not examined. Lastly, the changes in fibroblast growth factor 15, which is known to play important metabolic roles in response to FXR activation [41], could not be measured in this study due to technical difficulties.
In conclusion, this study demonstrated that the additional loss of FXR increases atherosclerosis and NAFLD characterized by macrovesicular hepatic steatosis in ApoE-deficient mice. These changes are associated with combined dyslipidemia due to increased fatty acid synthesis in liver, and increased adipocyte lipolysis. Treatment with a PPARα agonist reversed the deleterious effects of ectopic fat accumulation by decreasing plasma triglyceride levels associated with enhanced triglyceride hydrolysis and β-oxidation of fatty acids.
 XML Download
XML Download