

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Thyroid hormone (TH) deiodinases play a pivotal role in the functional diversification of TH signaling. They are involved in development, growth, and metabolic processes, and act in a cell-specific manner in the fine regulation of TH homeostasis. TH deiodinases catalyze activation and inactivation of THs through the removal of one iodine atom in the outer or inner ring of the TH molecule. They are expressed in virtually all organs and tissues, and a deiodinase is usually the initial or final actor in the fate of TH inside the cell. The major circulating TH is thyroxine (T4), whose synthesis is limited to the thyroid gland. T4 is considered a pro-hormone, which, at the local level, is regulated by tissue-specific ways by transformation into activated (triiodothyronine [T3]) or inactivated (reverse-T3 [rT3]) metabolites.
Three different deiodinases have been described: iodothyronine deiodinase 1 (DIO1), DIO2, and DIO3. These enzymes share important similarities despite being the products of different genes and having specific substrate affinities and physiological roles. Iodothyronine deiodination is considered to be the first step in the regulation of TH action, either in physiological or pathological conditions, wherein there is a need for diverse deiodinase distribution and function.
The present review summarizes the available information on the characteristics and functions of deiodinases. More specifically, we address the role of the three enzymes and their link to several pathological conditions in two of the main targets of THs, the heart and the brain. Furthermore, the impact of oxidative stress (OxS) on deiodinases and THs will be discussed.
GENERAL FEATURES OF DEIODINASES
Deiodinase genes
The three deiodinases are synthesized by three specific genes (DIO1, DIO2, and DIO3); however, due to the high sequence similarities, they probably derived from a common ancestral gene. The chromosome locations of the genes are different across species, and DIO1 and DIO3 are co-localized in most species. The size of introns varies among species, which is probably explainable by evolutionary reasons; interestingly, DIO3 does not contain introns, an exceedingly rare characteristic in the eukaryotic genome [1]. The DIO1 promoter has been particularly investigated in humans [2] and two specific thyroid response elements (TREs) have been identified at the 5′ flanking region of DIO1 gene, upstream with respect to the transcription start site. Both TREs functionally cooperate to ensure correct thyroid receptor binding, and specific mutations inactivating one of the two TREs block the T3 regulation of DIO1 expression [3]. Comparative studies on DIO2 indicate that, despite the high homology between rodent and human DIO2 promoters, their responsiveness to some transcription factors involved in thyroid metabolism is completely different. For example, the absence of responsiveness to thyroid transcription factor-1 (TTF-1) probably explains, at least in part, the lower expression of DIO2 in rat thyroid than in human thyroid [4]. DIO3 (both human and mouse) maps onto chromosomal imprinted regions, formed by a cluster of genes sharing the same regulatory element. This peculiarity makes it a possible candidate for phenotypic alterations associated with uniparental disomy of hosting chromosomes (human chromosome 14 and mouse chromosome 12) [5].
The subcellular location of deiodinases
Upon entering cells, THs can be activated or inactivated by deiodinases. The three deiodinases are dimeric integral membrane proteins of about 60 kDa, with a transmembrane domain involving the first 30 to 40 amino acids. They are characterized by the modified amino acid selenocysteine (SeCys) in the active center, which confers catalytic activity since at physiological pH, SeCys is ionized and acts as a very efficient electron donor, making the deiodination reaction possible [6]. The catalytic center contains a highly conservative core sequence that is important for dimerization, which is mediated by disulfide bonds [7]. Homodimerization remains the most frequent form of interaction between monomers of deiodinases; however, a certain level of DIO3:DIO1 and DIO3:DIO2 heterodimerization has been observed in living isolated cells for unclear reasons [8,9]. The three enzymes have different subcellular locations (Fig. 1); DIO1 and DIO3 are found at the plasma membrane, whereas DIO2 is found at the endoplasmic reticulum, making it very proximal to the nucleus. The DIO1 and DIO2 catalytic globular domains face the cytosol, whereas DIO3 molecules, including the catalytic domain, mostly protrude towards the extracellular space. DIO3 is rapidly internalized into cells through endosome vesicles and recycled back to the cell surface [10,11]. The cellular location of the three deiodinases is functionally associated with their role in maintaining the equilibrium with the plasma compartment (Fig. 1). The presence of DIO1 in the plasma membrane is associated with its major role in rapid equilibration with plasma T3, whereas the DIO2-mediated activation of TH is principally finalized in the nuclear compartment due to the location of DIO2 in the endoplasmic reticulum membrane, which is very proximal to the nucleus [10]. Even though immunohistochemical studies have localized the catalytic domain of DIO3 on the external side of plasma membrane, it was also colocalized with endosomal markers, suggesting that DIO3 is internalized by an endosome-mediated process. It has been hypothesized that the DIO3-catalyzed reaction may occur inside the endosome vesicles where THs are actively transported [11].
Deiodinase reactions, functions, and tissue distribution
Deiodination is the first step in the activation/inactivation process of THs and involves the removal of removes one iodine atom from the outer tyrosyl ring of T4 to produce T3, which is considered the biologically active form of TH with a short half-life of about 12 hours (whereas T4 has a half-life about 8 hours). DIO1 and DIO2 catalyze the outer ring deiodination of T4 and its conversion to active hormone T3, promoting the activation pathway [12]. The majority of circulating T3 derives from T4 deiodination by DIO1 activity, which mainly occurs in the thyroid, but also in the liver and kidney; typically, its presence is not evaluated (or is considered irrelevant) in other tissues. In contrast, DIO2 is considered as primarily responsible for the local production of T3 inside cells and its presence has been detected in several locations, such as the pituitary gland and hypothalamus [13,14], cochlea [15]), brown adipose tissue [16], bones [17], muscles [18], heart [19], and central nervous system [20]. Although the details underlying the distinction between DIO1-generated T3 and DIO2-generated T3 are still not completely known, it is clear that DIO1-generated T3 equilibrates rapidly with the plasma in about 30 minutes, whereas DIO2-generated T3 persists longer in the cell [5]. Furthermore, the finding that DIO2 is upregulated in patients with hypothyroidism and downregulated in those with hyperthyroidism suggests that changes in DIO2 contribute to the general effort of maintaining T3 homeostasis, both at circulating and tissue levels, in these two opposite pathological conditions [6]. Instead, DIO3 only catalyzes the inner ring deiodination of T4, resulting in the formation of the inactive product rT3. The enzymatic reaction of the three deiodinases depends on the presence of SeCys in the active catalytic center, and the substitution of this amino acid with Cys seriously reduces (by 2 to 3 orders of magnitude) the affinity of deiodinases for their specific preferred substrate. This effect is particularly evident in the reduction of the affinity of DIO2 for T4 and that of DIO3 for T3. The affinity of DIO1 for rT3 is less strongly influenced by the SeCys-Cys substitution [21].
DIO1 was the first deiodinase to be identified and cloned. It catalyzes deiodination of both the inner ring of T4 (T4 to rT3) and the outer ring of T4 (T4 to T3) with the same efficacy, as shown by the comparable Vmax/Km ratios for the two reactions (9 to 13 μM·min/pmol·mg protein). The DIO1 reaction follows a ping-pong kinetic pattern in which two substrates, the TH and a thiol cofactor, compete for interaction with the enzyme. This reaction is inhibited by 6n-propylthiouracil (PTU), which competes with the cofactor and irreversibly links the enzyme to an inactivated complex [21]. In contrast, DIO2 and DIO3 have sequential kinetics, wherein the two substrates interact with the enzyme at the same time. DIO2 is an outer ring deiodinase and T4 is considered to be its natural substrate. DIO2 has a Km for T4 in the nanomolar range, requires a thiol cofactor, and is not as sensitive as DIO1 to PTU. DIO2 activity is regulated at both transcriptional and posttranslational levels and seems to involve the proteasome system [22,23]. The observation at several cell and tissue levels that exposure to TH induces a marked reduction of DIO2 activity, but no change in DIO2 mRNA, strongly supports a posttranslational mechanism of regulation [24]. The variability of the DIO2 activity/mRNA ratio is further explained by the short half-life of the enzyme (about 40 minutes), which is reduced after exposure to T4, through a regulatory feedback loop that regulates the intracellular production and availability of T3. Selective degradation by proteosomes requires, as the first step, the conjugation of the protein to a small conserved protein called ubiquitin, which triggers the proteasome enzymatic cascade that inactivates DIO2 by disrupting DIO2 homodimers [23,25,26]. DIO3 is an obligatory inner-ring deiodinase with half-life of 12 hours, which plays a fundamental role in the inactivation of circulating TH production. DIO3 catalyzes the conversion of T4 to rT3 and T3 to 3,3′-diiodothyronine (T2), both of which are biologically inactive. DIO3 is the main deiodinase during the embryonic period and in developing tissues, protecting developing tissues from excessive TH production. In adulthood, DIO3 is mainly present in the central nervous system [27] and skin [28], while most other tissues show very little or no DIO3 expression in adulthood under physiological conditions. Furthermore, the high expression of DIO3 in the placenta and pregnant uterus suggests that this enzyme plays an important role in the development process [29]. The protective role of this enzyme in adulthood has been confirmed only in cases of TH overloading observed in some critical illnesses associated with hypoxic/ischemic conditions [30]. The main characteristics of the three deiodinases are presented in Table 1.
DEIODINASES AND TH SIGNALING
In euthyroid adults, circulating T4 is converted to T3 upon entering deiodinase-containing cells, which are widely distributed throughout the body. New T3 molecules return to the circulation, adding to the pool directly secreted by the thyroid gland. The majority of T3 produced in the body derives from DIO2 activity, which has been found in many tissues. The first step in TH action is the binding of available intracellular T3 to TH receptors (TRs): TRα (TRα1 and TRα2) and TRβ (TRβ1 and TRβ2). In euthyroid conditions, it has been estimated that in the liver and kidney, about 50% of TRs are occupied by circulating T3 entering the cells [5]. In pathological conditions the ratio between TRs and TH may fluctuate markedly. Virtually all tissues in the body receive the same signal from circulating T3; however, the biological response is quite different since it also depends on the local activity of DIO2, which acts as an additional source of cellular T3, in accordance with the metabolic requirements of the cells and tissue. The synergistic action of deiodinases is crucial for the availability of TH in the cell, despite the level of T3 entering the cell. For example, in the mouse brain, T3 levels in some areas of the brain drop by about 50% after selective disruption of the Dio2 gene, and this finding was further confirmed by the increase of T4 in the brains of these Dio2-knockout (KO) mice [31]. Moreover, cold exposure stimulates DIO2 expression and increases T3 levels in brown adipose tissue, which adds to the T3 pool entering from the circulation. This process results in the massive increase of intracellular T3, TR saturation, and T3-dependent gene expression [19]. Similar DIO2-mediated increases in T3 have been observed in many other organs, such as the brain, liver, and skeletal muscle [32,33].
In contrast, DIO3-expressing tissues are characterized by low levels of T3 and a gene expression profile typical of hypothyroid conditions since DIO3 inactivates both T4 and T3 at the plasma membrane level as soon as they enter the cell, even before any interactions with TRs [34]. The general properties of deiodinases allow them to work as a powerful customized mechanism regulating TH signaling, which depends on the tissue pathophysiological conditions, metabolic imprinting, and functional requirements.
Deiodinases and the heart: low T3 syndrome and pathological remodeling
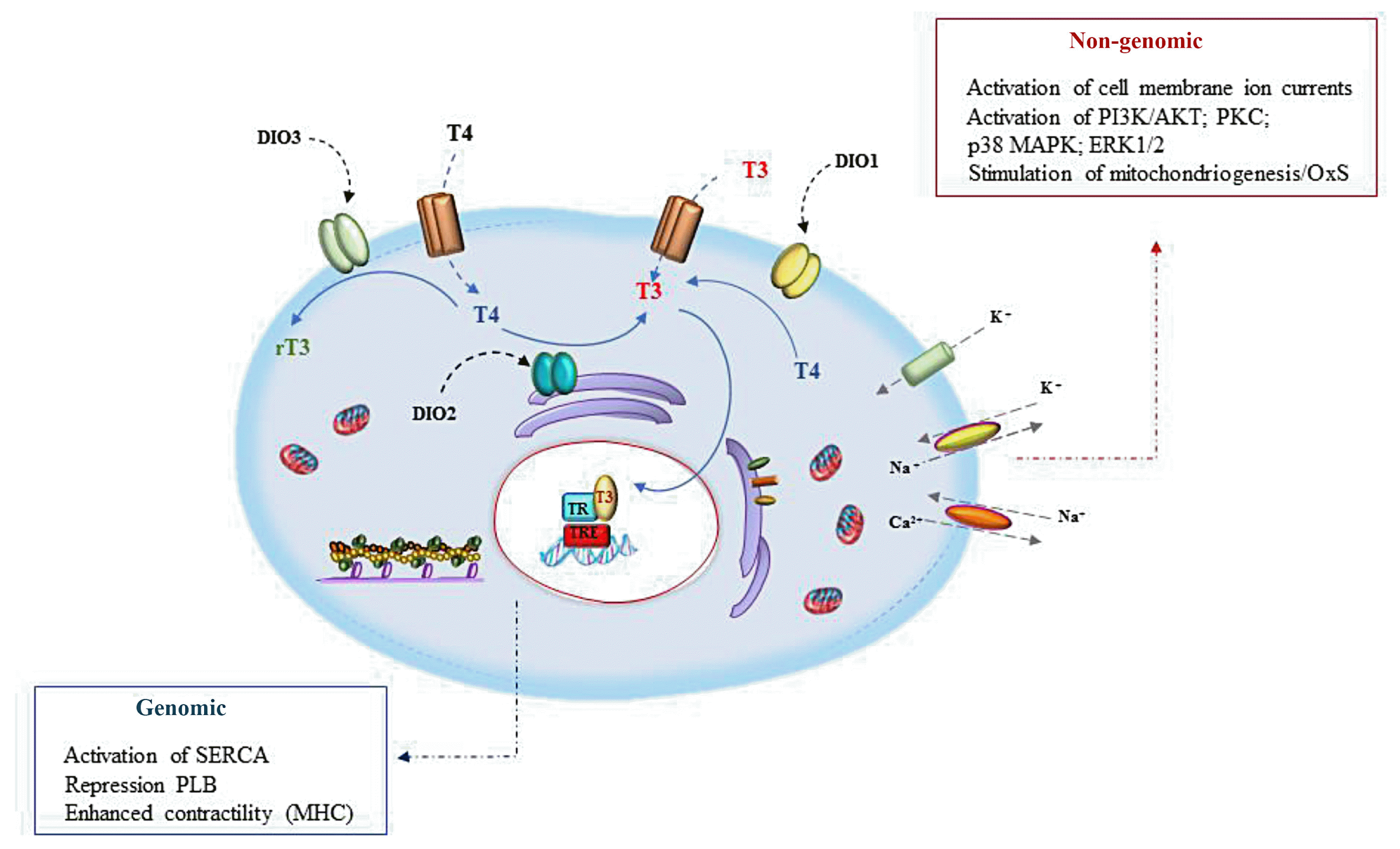
THs play a central role in the heart, and changes in circulating or tissue TH levels are associated with important modifications of heart metabolism and function by genomic and non-genomic effects (Fig. 1) [35].
Genomic effects depend on the transcriptional regulation of specific genes and require the nuclear uptake of T3, T3-TR complex formation, and the successive occupation of specific TREs in the target gene promoter. The direct effects of TH on cardiac muscle are principally associated with the regulation of myosin heavy chain (MHC) α and β. The regulation of cardiac contractility by TH also occurs through positive regulation of the calcium ATPase protein in the sarcoplasmic reticulum (sarco/endoplasmic reticulum Ca2+ adenosine triphosphatase-2a [SERCA2a]) and downregulation by phospholamban (PLB) [36]. SERCA2 pumps calcium ions in the sarcoplasmic reticulum inducing relaxation of myofibrils, whereas PLB functionally counteracts this activity.
The non-genomic effects of THs do not involve the cell nucleus and are principally described as occurring in the mitochondria and cytoskeleton [37]. The non-genomic actions of TH are rapid (minutes or a few hours) and initiate at receptors present at the cell membrane (e.g., integrin ανβ3) or cytoplasm (e.g., truncated TRα) and do not require gene transcription and translation [38]. In different cell types, in response to THs, various cell signaling pathways are activated, including the mitogen-activated protein kinase (MAPK) superfamily (extracellular signal-regulated kinases [ERK1/2]; p30 MAPK; protein kinase C [PKC], and phosphoinositide 3-kinases [PI3K]/Akt) [38].
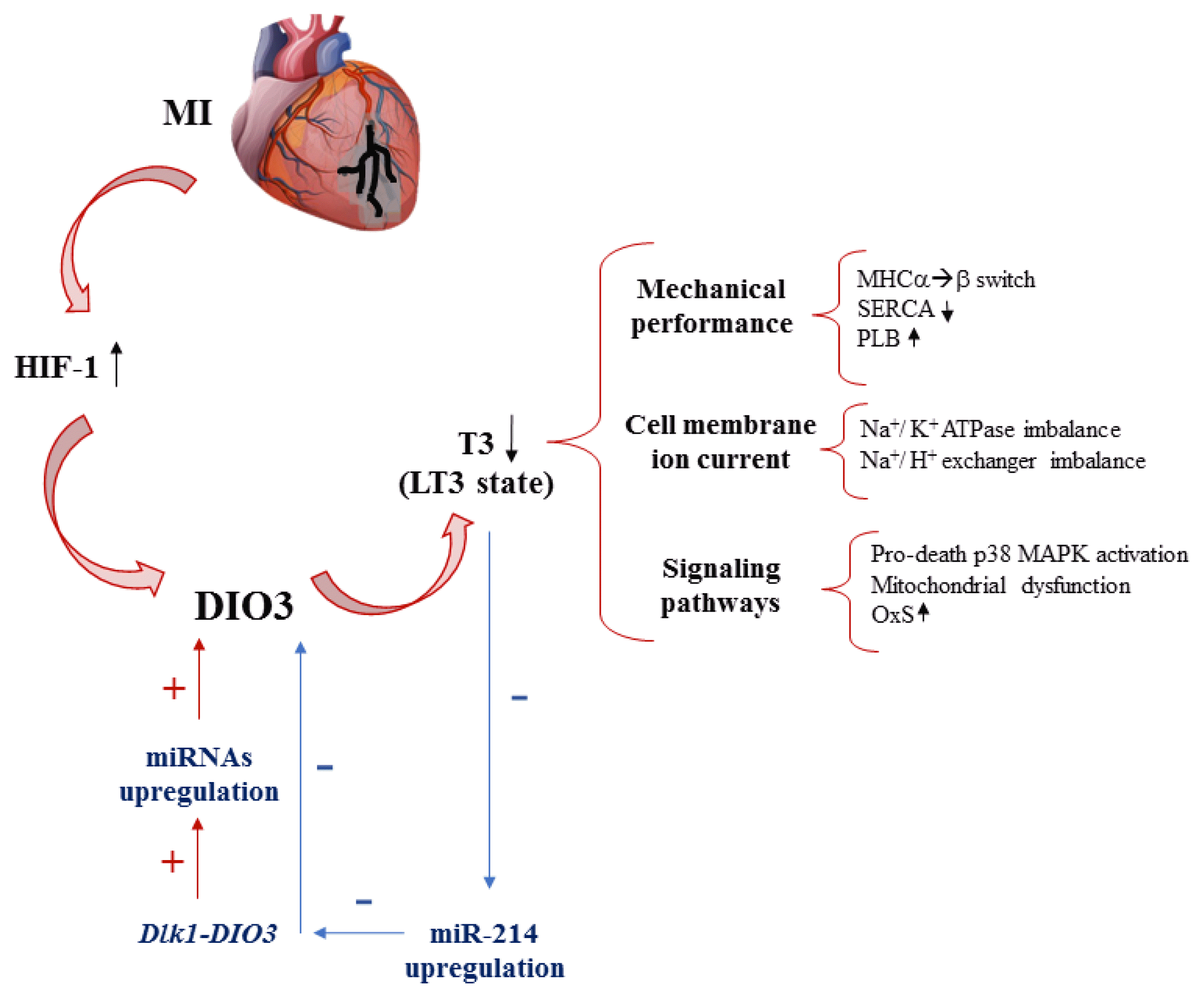
Interactions between TH and the heart are particularly evident in low T3 (LT3) syndrome, when severe systemic illnesses, including heart failure and myocardial infarction (MI), lead to alteration of the functions of TH. LT3 syndrome is characterized by a fall in serum T3 levels that may be accompanied by a drop in circulating T4. Serum thyrotropin levels are usually normal, but may be slightly increased or even decreased [39]. At the molecular level, the T4 drop associated with LT3 syndrome is attributed mainly to DIO3 activity, which inactivates T4 to rT3 and T2 [40]. A reduction of oxygen availability in myocardial ischemia and associated hypoxia is considered to be main cause of significant augmented DIO3 neoexpression. DIO3, in fact, is a direct target of hypoxia-inducible factor 1 (HIF-1), and persistent HIF-1 signaling is associated with the hypertrophic remodeling of surviving cardiac tissue [34]. Impairment of TH signaling reduces energy turnover and promotes contractile dysfunction, leading the heart to pathological remodeling and, ultimately, to heart failure.
Deiodinase activation in pathological myocardium was observed for the first time in ischemic human heart tissue and in rodent models of cardiac hypertrophy and failure [40–42]. The re-expression of the so-called “fetal gene program,” which is associated with cardiac remodeling and involving many of the genes expressed during the fetal life, can be seen as a compensatory attempt of the pathological heart to maintain the cardiac output in the most efficient manner possible, reducing energy expenditure and T3 availability (Fig. 2). However, this initial adaptive response ultimately becomes maladaptive because of the progressive decline in cardiac function [43]. DIO3 is one of the main genes of the fetal program reactivation, and an increase in its expression correlates with important changes in cardiac gene expression associated with T3, including the switch from the MHCα isoform to the MHCβ and the reduction of SERCA activity with the impairment of calcium uptake in the endoplasmic reticulum [44]. Recently, in a rat model of ischemia, after reperfusion of overt LT3 animals with physiological levels of T3, the three deiodinases showed different expression profiles depending on the proximity of the analyzed tissue to the ischemic event. The global T3-mediated amelioration of cardiac function and structure suggests a customized role of the effects of the three deiodinases during the ischemic reperfusion process in order to restore efficient intracellular TH levels to the extent possible, thus opposing the LT3 condition. A further insight emerging from this study is the existence of regional regulation of deiodinases in different cardiac areas on the basis of tissue insults [45]. However, even though DIO3 is considered to be one of the undisputed protagonists in the development of heart failure, the mechanisms of its regulation are still largely incompletely understood. From this perspective, the evaluation of the possible involvement of microRNAs (miRNAs) in the regulation of DIO3 mRNA expression was investigated. MiRNAs are small non-coding RNA molecules composed of about 22 nucleotides that post-transcriptionally regulate protein synthesis by binding to target mRNAs [46]. Alterations in miRNA-biogenesis pathways have been demonstrated to be linked with myocyte hypertrophy, cardiac fibrosis, and fetal gene reprogramming, suggesting a central role of miRNAs in cardiac remodeling and the development of heart failure. An analysis of the miRNA expression profile in the left ventricle of a MI mouse model showed that a set of 29 upregulated miRNAs originating from the delta-like homolog 1-DIO3 (Dlk1)-Dio3 region were closely associated with pluripotency and proliferation activities, particularly in tumors and the progression of several diseases [47,48]. Interestingly, this miRNA signature was associated with significant induction of DIO3 expression in the left ventricle of MI rats, suggesting the possible involvement of these miRNAs in the compensatory process activated in the injured myocardium and mediated by DIO3 reactivation [48]. Furthermore, data from a MI mouse model and an in vitro analysis indicated that cardiac miR-214 has direct functional interactions with DIO3 mRNA and its target site is located in the “selenocysteine insertion sequence” (SECIS), responsible for the incorporation of SeCys in the active catalytic center. The reduction of cardiac T3 induces miR-214, which exerts inhibitory effects on DIO3 expression in an attempt to restore T3 levels. Therefore, miR-214 has a protective function on infarcted myocardium (Fig. 2) [49].
Among the transcriptional and signaling pathways involved in the regulation of pathological cardiac remodeling, recently, the forkhead box O (FoxO) family of transcription factors has emerged as important players as stress inducers. FoxO factors have a conserved 110-amino-acid DNA-binding motif called the “forkhead box” [50,51]. In particular, the forkhead domain of four members of the group (FoxO1, FoxO3, FoxO4, and FoxO6) recognizes a consensus DNA-binding element in numerous genes, called the FoxO-responsive element. Different FoxO factors have distinct roles in cardiac remodeling, and their mechanism of action is far from being elucidated. Among FoxO factors, FoxO1 has been reported to be directly involved in cardiovascular morphogenesis and metabolic stress-induced cardiac remodeling. Interestingly, DIO2 is reported as a direct downstream target of FoxO1 in cardiomyocytes, and a FoxO1-dependent reciprocal regulation of DIO2 and DIO3 expression was also observed as a regulatory step in hypertrophic cardiomyocyte growth and pathological remodeling of the heart [52].
Collectively, all data heretofore reported on the heart suggest that in response to severe insults, DIO2 and DIO3 expression, most of the time in a reciprocal fashion, modify TH signaling in localized organ areas, which could affect the balance between adaptive and maladaptive mechanisms. Therefore, TH signaling reduction during hypoxic-ischemic events reduces energy expenditure and oxygen consumption, and it could be interpreted as an adaptive mechanism. From this perspective, the potential role of TH in therapies addressing the molecular biology of TH-metabolism components is an extremely promising challenge for which multidisciplinary research approaches may be suitable.
Deiodinases and the brain: dynamic regulation
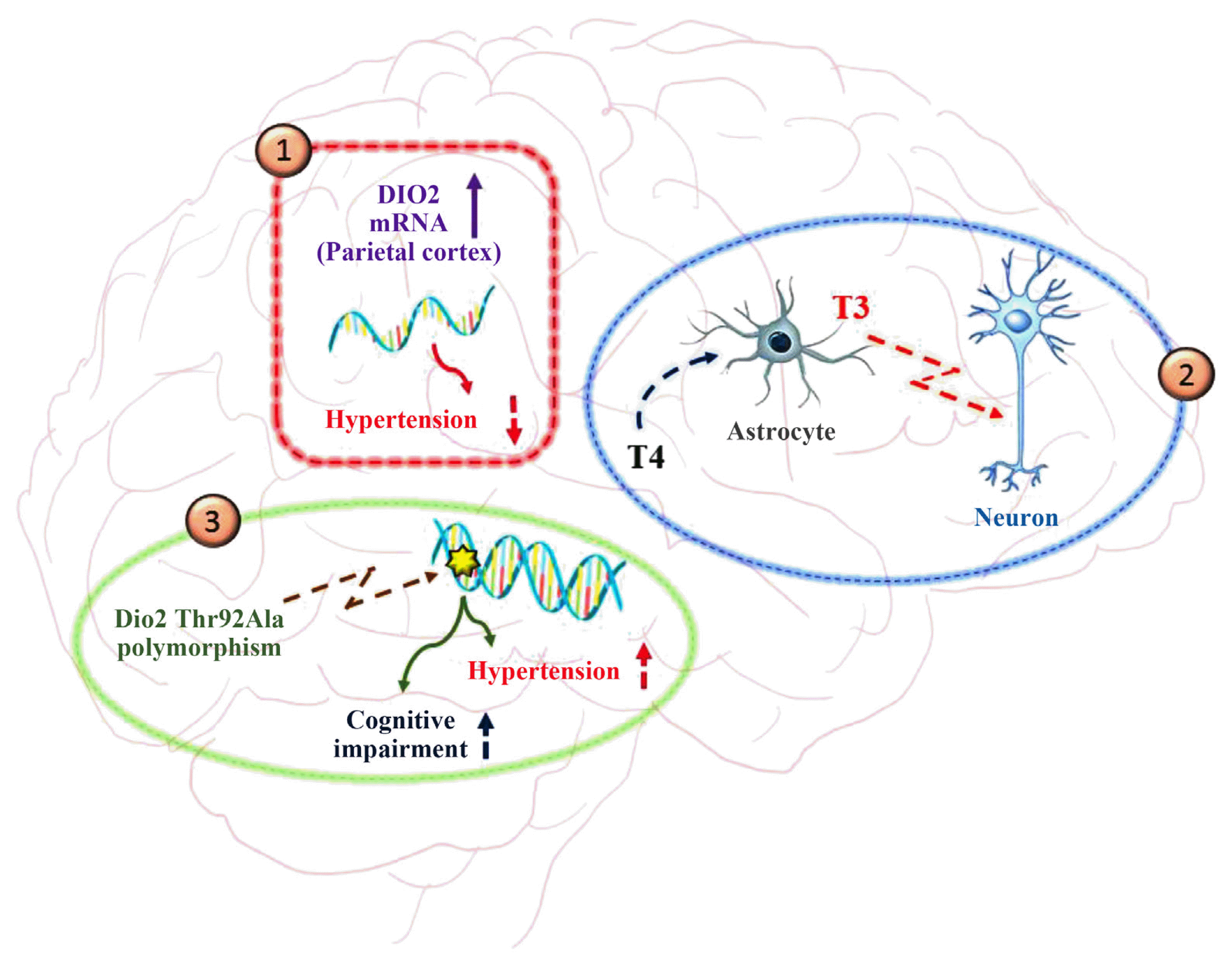
In the last decade, the functional role of TH in the brain has attracted considerable interest. It is well known that TH is crucial in brain development since they are involved in neural cell migration, differentiation, and signaling [30]. Rodent brain studies have identified many genes regulated by TH [30]. Activation of TH has been observed mainly in glial cells, astrocytes, and tanycytes, while the principal target cells are neurons and oligodendrocytes. TH exerts regulatory functions on gene expression involved in myelination, cell differentiation, migration, and signaling [53,54]. Most T3 in the brain is locally produced by DIO2 through paracrine-mediated signaling, in which a regulatory pathway proceeds from DIO2 in glial cells and TRs in neurons, establishing deiodinases as a checkpoint for the regulation of TH signaling in the brain in health and disease (Fig. 3) [45]. Studies on Dio2-KO mice showed that even though serum T3 levels were normal, T3 levels in the brain were greatly reduced, probably because of a reduction in local T4 to T3 deiodination. Since no augmented DIO3 activity was observed in various areas of the Dio2-KO mouse brain, we can assume that T3 reduction was not attributable to an increase in its degradation. Moreover, the fact that T3 was still present in cells, albeit at a lower concentration, suggests that the brain is not completely dependent on local production of T3 by DIO2 and that a considerable amount of the hormone is derived from the serum and cerebrospinal fluid. This mitigates neurological damage that could result from a deficiency of DIO2. A completely different situation can be observed in hypothyroid mice, where both serum T4 and T3 levels were reduced and higher DIO2 activity was observed in an attempt to compensate for the lack of hormones [31].
The homeostatic efficiency of deiodinases is particularly evident in cases of iodine deficiency, where serum levels of T3 are preserved by a potentiation of DIO2 activity and reduction of T3 clearance by DIO3. However, in such conditions, TH signaling is endangered in tissues such as the brain, strictly depending on local DIO2-mediated T3 production. In these contexts, fine-tuned regulation of DIO2 and DIO3 expression and DIO2 and DIO3 activity is observed, since iodine deficiency inhibits DIO3 expression and DIO3 activity, whereas DIO2 is greatly potentiated [55].
Recently, the relevance of deiodinase triad homeostasis in the brain has been highlighted in a model of hypertension, where the presence of the three deiodinases was evaluated in frontal and parietal cortex of spontaneously hypertensive rats (SHRs). Interestingly, Dio1, Dio2, and Dio3 mRNA was expressed in both regions, but only Dio2 mRNA expression was significantly increased in the frontal cortex after a mechanical hypotensive stimulus, induced by two successive mandibular extensions, suggesting a major involvement of this enzyme in an attempt to restore more physiological conditions and correct T3 levels, associated with normotensive status in the brain (Fig. 3). Moreover, the same study showed higher DIO1 protein levels in the parietal (but not frontal) cortex of SHR brain after two mandibular extensions, raising interesting questions about the role of this enzyme in TH system homeostasis in hypertensive conditions, which await further investigations [56].
Another important field of investigation dealing with deiodinases in the brain regards the description of several DIO gene-associated polymorphisms with important physiological roles. As an example, the DIO1 Cys785Thr polymorphism correlates with a reduction of circulating T3 and an increase of T4 and rT3 levels, thus indicating that it acts on DIO1 activity reduction. The opposite happens when Ala1814Gly is present on DIO1 and an increase of DIO1 activity can be described [57]. In particular, the DIO2 Thr92Ala is the most studied polymorphism in human brain and cell models, given its high recurrence (about 12% to 36% of homozygosity) in the human population. The DIO2 variant is less efficient than the wild type and can also be accumulated in the Golgi apparatus [58]. The expression of DIO2 Thr92Ala interferes with cellular processes first by damaging the endoplasmic reticulum and Golgi apparatus and then promoting mitochondrial unbalancing, inflammation, and apoptosis. DIO2 Thr92Ala in the brain is closely correlated to impaired cognition, and the fact that short-term treatment with T3 reduces behavioral impairment indicates the presence of hypothyroid areas in the brain carrying this polymorphism (Fig. 3) [59]. New investigations are needed to better understand the functional link between the TH system and cognitive imbalance and the role of DIO2 and its variants on this occurrence.
The DIO2 Thr92Ala mutation has also been found to increase hypertension susceptibility, further augmenting interest in the role of TH in the pathogenesis of hypertension [60]. The presence of DIO2 in vascular muscle cells reflects its role in vascular tone regulation [61]. The observation of a stronger association between DIO2 Thr92Ala and serum thyroid-stimulating hormone levels in individuals with hypertension suggests that this DIO2 variant may serve as a phenotypic marker of hypertension [60]. Based on these results, with the goal of better defining the functional link between the TH system and hypertension in different regions of the brain, the availability of appropriate hypertensive rat models (such as the previously mentioned SHRs) will help to understand the involvement of Dio2 genetic variants in the progression of hypertension.
In summary, given the fundamental role of deiodinases in the assessment of correct T3 concentrations in the brain, the study of DIO gene-associated polymorphisms may add important information on the molecular mechanisms leading to different pathological conditions. Consequently, from a clinical perspective, the data obtained can potentially help to individuate patients with a higher risk for poor outcomes.
Deiodinases, THs, and OxS
THs have a consistent impact on whole-body energy metabolism and tissue-specific energy balance. For this reason, altered TH concentrations are associated with profound changes in energy status [62]. At the intracellular level, TH levels are under the control of deiodinases that catalyze TH activation or catabolism, through cell-specific dynamics, often regardless of constant serum T3 levels [5].
It is well known that changes in cellular OxS induce an increase of DIO3 activity in different disease states, leading to alterations in TH metabolism [63,64]. Conversely, DIO1 and DIO2 activities appear to be reduced, with a consequent decrease in serum T3 levels and increased rT3 concentrations [65]. Hypoxia, as is elicited during MI, upregulates DIO3, which is negatively correlated with T3 circulating levels [34,63]. The progressive worsening of cardiac function has been associated with compromised left ventricular geometry and pathological remodeling in several rat models of cardiac ischemia and human MI patients [34,64,66]. Conversely, other experimental and clinical studies demonstrated that antioxidant supplementation after MI can rebalance the redox status, preventing alterations in TH levels and improving cardiac function [67]. In an experimental model of MI rats, T3 has been demonstrated to reduce reactive oxygen species (ROS) levels, increase endothelial nitric oxide synthase expression, and improve nitric oxide availability [68]. Moreover, T3 is downregulated in arsenic-induced testicular oxidative damage in rats, in parallel with a fall in superoxide dismutase (SOD), catalase, glutathione (GSH), and zinc levels, and the increase of malondialdehyde (MDA) and tumor necrosis factor-alpha [69]. However, other results have shown that T3 reduced ROS and MDA levels but enhanced SOD and GSH-peroxidase levels in a model of oxygen-glucose deprived cardiomyocytes characterized by a significant upregulation of ROS and MDA and downregulation of SOD and GSH-peroxidase [67]. Other data confirmed that TH administration might reduce OxS, modulate mitochondrial activity, and protect against lipid peroxidation [70–72]. Taken together, these results suggest that T3 protects cells from OxS by improving antioxidant levels and reducing the levels of their oxidant counterparts.
Inflammation fits into the relationship between deiodinases, T3, and OxS, with particularly substantial data found for interleukin 6 (IL-6). In fact, higher IL-6 levels increase superoxide radical generation through the nicotinammide adenina dinucleotide fosfato (NAD(P)H) oxidase system, impair DIO1 and DIO2 function, and induce the expression of DIO3 [73,74].
Given the importance of THs, several approaches have been attempted to prevent the deleterious effects of T3 inactivation by antioxidant treatment. Indeed, N-acetylcysteine (NAC), a thiol-containing radical scavenger that restores reduced intracellular GSH levels, prevents IL6-induced effects on the intracellular redox state, and reduces DIO3 expression. Although the effect of NAC on DIO3 expression and activity is still not fully understood, it likely involves the maintenance of intracellular thiols, such as GSH, as well as the modulation of redox balance [74].
A critical molecule in counteracting intracellular OxS is selenium, an essential trace element that is a component of selenoproteins, which have antioxidant properties [75]. Selenium plays a crucial role in TH metabolism due to its location in the catalytic site of the three deiodinases [76]. Since the thyroid gland, with its capacity to store selenium, maintains a high concentration of this element, it is not easy to evaluate the immediate benefits of dietary selenium supplementation on selenium-containing enzymes [77]. Nonetheless, clinical data suggest that selenium deficiency is associated with an increased risk of a high anti-thyroid antibody titer, whereas selenium supplementation decreases thyroid peroxidase antibody titer, which could allow routine selenium supplementation as an additive tool to permit a reduction in the levothyroxine dose required for hypothyroidism therapy or to prevent progression of subclinical hypothyroidism [78–80].
In a cell culture model, the addition of sodium selenite partially corrected the augmented ROS levels (reduction between 30% and 50%) and protein damage (carbonyl group reduction by 50% to 60%) induced by elevated IL-6 levels [75]. Variations in the intracellular redox state may seriously damage the function of deiodinases since they are oxidoreductases. Against all expectations, sodium selenite supplementation did not seem able to restore DIO1 and DIO2 functions. A possible explanation may relate to the depleted amount of thiol cofactors due to OxS and the consequent impairment of the deiodinase reaction. In contrast, sodium selenite influences IL-6-induced DIO3 activation, and the specific location of the enzyme may play a role in making this enzyme less accessible [75].
Interestingly, correction of the redox state (decreased carbonyl levels) and a reduction in DIO3 activity in only 4 weeks of exercise training was previously observed in an experimental model of MI, resulting in improvements in cardiac function in terms of left ventricular mass, cardiac output, and the ejection fraction (assessed by echocardiography). A negative correlation of DIO3 with the maximum effort capacity (evaluated by an exercise stress test) was also found [81]. Thus, it is possible to consider exercise as a non-pharmacological tool to help to restore antioxidant defenses and T3 biological activity [82,83]. Changes in THs vary considerably according to the type, intensity, and duration of exercise, showing increases, decreases, or a lack of changes, and in different subject cohorts, depending on tissue-specific enzymatic activities [84]. Thus, this interesting issue requires a deeper analysis to understand the effects of exercise on TH function and OxS, and how different populations may have benefits in terms of health and safety.
CONCLUSIONS
In conclusion, TH deiodinases constitute a dynamic system whose components act in a synergistic manner with the final aim of maintaining TH signaling unchanged, to the extent possible, in the serum and in the intracellular environment, thereby supporting thyroid function in relation to various demands of the organism in physiological and pathological contexts. Given the relevance of the thyroid system in all aspects of metabolism, an understanding of mechanisms driving deiodinase homeostasis may have a fundamental impact on adequate therapeutical approaches.
 XML Download
XML Download