

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Papillary thyroid carcinoma (PTC) is the most common endocrine malignancy and accounts for nearly 90% of all thyroid cancers [1,2]. The rising incidence rate of PTC may be a result of either an increase in the occurrence of this disease or a continuous improvement of its diagnosis [3]. PTC frequently shows local lymph node metastasis or rarely distant metastasis to the lung or bone [4]. Most PTC patients have a good prognosis, with aggressive surgical resection and radioiodine (RI) remnant ablation; however, these effective managements are limited to resectable or RI-avid metastatic PTC [5]. Therefore, there is an unmet need for new therapeutic drugs for RI-refractory thyroid cancers.
Epithelial-mesenchymal transition (EMT) is a process during which epithelial cells lose their original polarity and are transformed to a mesenchymal phenotype [6,7]. During EMT, cancer cells acquire potent growth signals to obtain invasive capacity, breakdown the basement membrane, and activate the largest process of metastasis [8]. EMT is also defined by the loss of cell-cell adhesion, regulation of cell-surface proteins such as E-cadherin and N-cadherin, induction of cytoskeletal proteins such as Vimentin, and increase the expression of transcription factors such as Snail [9]. Transforming growth factor-beta1 (TGF-β1) is a cytokine belonging to the transforming growth factor superfamily, which has multiple functions and plays a leading role in inducing EMT during cancer metastasis [10].
TGF-β1 was overexpressed in PTC compared to normal thyroid tissue and high level of TGF-β1 mRNA was detected in the serum of patients with PTC compared with in healthy controls [11]. TGF-β1 plays a major role in promoting tumor invasion during PTC progression [12]. Thyroid cancer from BRAFV600E transgenic mice was susceptible to TGF-β1 induced EMT [13]. In terms of signaling pathways, non-Smad pathways such as mitogen-activated protein kinase (MAPK), Rho, and phosphoinositide 3-kinase (PI3K) signaling, as well as the conventional Smad pathway, were also involved in TGF-β1-induced EMT [14]. Previous studies have demonstrated that TGF-β1 mediated tumor cell EMT and metastasis through the MAPK pathway [15]. Furthermore, tumor cells can migrate and invade target organs by expressing matrix metalloproteinases (MMPs), such as MMP-2, to degrade the extracellular matrix (ECM) proteins [16]. These studies may reveal that clarifying the relationship between EMT and MAPK signaling pathways may play a crucial role in PTC metastasis.
Increasing evidences of epidemiological, and clinical studies have suggested a link between diabetes and cancer, since patients with diabetes have greater risk of thyroid malignancy [17]. Multiple risk factors including hyperinsulinemia, hyperlipidemia, hyperglycemia, and inflammation potentially contribute to progression of cancer in type 2 diabetes [18]. Earlier clinical study demonstrated that one antidiabetic drug, rosiglitazone (RGZ) used in patients reduce the risk of thyroid cancer [19].
Previous studies have shown that peroxisome proliferator-activated receptor gamma (PPAR-γ) ligands, such as RGZ, troglitazone, and pioglitazone can inhibit cancer cell proliferation and metastasis in lung, breast, and pancreatic cancers both in vitro and in vivo [20–22]. One earlier clinical study proposed the possible antitumor effects of pioglitazone against metastatic paired box 8 (PAX8)-PPAR-γ fusion protein thyroid carcinoma [23]. Early animal study showed that RGZ effectively inhibit the progression of thyroid carcinogenesis in mouse model [24]. Moreover, it was shown that RGZ induced a partial reversion of EMT in anaplastic thyroid cancer (ATC) cells by multiple mechanisms [25]. All these studies suggest that PPAR-γ agonists have a potential antitumor role in thyroid cancer treatment.
Lobeglitazone (LGZ), a novel activator of PPAR-γ, is a widely used thiazolidinedione (TZD) for the treatment of type 2 diabetes in Korea [26]. Compared to other TZDs (RGZ and troglitazone), LGZ had an improved safety profile and did not contribute to bladder cancer tumorgenesis [27,28]. However, the anti-tumor effects of LGZ in thyroid cancer have not yet been recognized. In this study, we aimed to show that a new PPAR-γ ligand, LGZ, can prevent TGF-β1-induced cancer progression and metastasis in the most prevalent thyroid cancer, BRAF-mutated PTC cells.
METHODS
Cell culture and reagents
PTC cell lines (BCPAP and K1 were both harboring the BRAFV600E mutation) were grown in Roswell Park Memorial Institute 1640 (RPMI1640) or Dulbecco’s Modified Eagle Medium (DMEM)/F12 (1:1) medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and both were supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) at 37°C with 5% CO2. Recombinant human TGF-β1 (7754-BH, R&D systems, Minneapolis, MN, USA) was reconstituted at 100 μg/mL in sterile 4 mM HCl containing at least 0.1% bovine serum albumin (BSA). LGZ was provided by Chong Kun Dang Pharm. Inc, and another conventional PPAR-γ ligand, RGZ (122320-73-4, Sigma-Aldrich, St. Louis, MO, USA), and PPAR-γ antagonist-2-chloro-5-nitrobenzanilide (GW9662) (M6191, Sigma-Aldrich) were purchased and dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C. This study followed Institutional Review Board of Seoul St. Mary’s Hospital, The Catholic University of Korea, which indicated for exemption (Number: KC-20EISI0390).
Cellular viability assay
The viability of PTC cells was analyzed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, BCPAP and K1 cells were seeded into a 96-well cell culture plate at a density of 3×103 cells/well in 100 μL of growth media. Following 24 hours incubation at 37°C, cells were treated with LGZ (0.01 to 1,000 μM) or vehicle (DMSO) control and cultured at 37°C for 24 or 48 hours. MTT (298-93-1, ALL for LAB, Seoul, Korea), a stock solution of 5 mg/mL was added to each well and incubated at 37°C for 4 hours. The supernatant was removed. Subsequently, in order to dissolve the formazan crystals, 150 μL of DMSO was added to each well. The plate was shaken for 15 minutes at room temperature (RT). The absorbance was read at 540 nm using an ELISA reader (BioTek, Winooski, VT, USA).
Western bolt
Cells were washed twice with phosphate buffered saline (PBS) at 4°C and lysed in RIPA buffer. Protein levels in the cell lysates were measured using a standard Bradford assay. Total protein amount (10 μg) from each sample was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with BSA in Tris-buffered saline, 0.1% Tween® 20 Detergent (TBST) for 2 hours at RT, the membranes were incubated overnight at 4°C with specific primary antibodies: PPAR-γ (1:1,000; sc-7273; Santa Cruz Biotechnology, Dallas, TX, USA), CCAAT/enhancer binding protein alpha (C/EBPα; 1:500; D56F10, Cell Signaling Technology, Danvers, MA, USA), E-cadherin (1:1,000; ab40772, Abcam, Cambridge, UK), N-cadherin (1:1,000; 13116S, Cell Signaling Technology), Vimentin (1:1,000; 5741S, Cell Signaling Technology), Snail (1:1,000; Novus, NBP2-27293, Littleton, CO, USA), p-P38 (1:1,000; 4511, Novus), total-P38 (1:1,000; 9212, Novus), MMP-2 (1:1,000; GTX-104577, Genetex, Irvine, CA, USA), and β-actin (1:1,000; ab8227, Abcam). The membranes were then washed and incubated with the secondary antibodies goat anti-rabbit immunoglobulin G (IgG) or goat anti-mouse IgG (1:2,000; Cell Signaling Technology) for 2 hours with BSA in TBST. The bands were detected using an image analyzer system (Syngene, Cambridge, UK).
Immunocytochemistry
BCPAP and K1 cells were trypsinized and seeded on coverslips at 70% confluence. Cells were washed twice with PBS and fixed in 4% formaldehyde in PBS for 10 minutes at RT. The cells were permeabilized with 0.1% TritonX-100 for 60 minutes at RT. The cells were washed three times with PBS. Subsequently, the cells were blocked with 5% normal goat serum in PBS for 3 hours at RT. Fixed cells were incubated with the primary antibody E-cadherin overnight at 4°C. After washing with PBS three times, cells were incubated with secondary antibody Alexa Fluor 488 rabbit anti-rabbit IgG (A-21206, 1:500, Invitrogen) for 45 minutes at RT protected from light. Finally, the cells were washed with PBS three times and washed once with distilled water. The coverslips were mounted with a mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; AR-6501-01, ImmunoBioscience Corp., Mukilteo, WA, USA). Images were captured using a confocal laser scanning microscope LMS800 (Carl Zeiss AG, Oberkochen, Germany) at 640× magnification.
Cell migration assay
Cell suspensions containing 4×105 cells/mL were seeded on a 6-well plate. After 24 hours of incubation, the cells in each well reached 100% confluence, and a wound was made by using a sterile 200-μL pipette tip on the cell monolayer. Cells were washed with PBS until there were no floating cells and then serum-free medium was added. Cells were pretreated with the indicated concentrations of RGZ and LGZ for 30 minutes and then stimulated with TGF-β1 (20 ng/mL) for 24 hours. Images were captured using a Nikon Nikon-TS100 microscope (Nikon, Tokyo, Japan). The percentage scratch closure was calculated using ImageJ software.
Matrigel invasion assay
Matrigel invasion assays were performed according to the protocol of manufacturer’s instructions (Corning, Acton, MA, USA). A 100-μL of cell suspension (4×104 cells) in serum-free media was added to the upper chamber, while the lower chamber was filled with RPMI or DMEM containing 10% FBS. Each upper and lower chamber was separated by an 8-μm porous polycarbonate membrane. Cells were pretreated with LGZ and RGZ for 15 minutes in the upper chamber and then stimulated by the presence or absence of TGF-β1 (20 ng/mL) for 24 hours and incubated at 37°C with 5% CO2. After the medium was discarded, the cells were washed three times with PBS. Cells at the upper surface of the membrane were removed using a cotton swab. The invaded cells on the bottom of the membrane were fixed with 4% formaldehyde for 30 minutes and stained with hematoxylin and eosin (H&E) protected from light for 20 minutes. For each filter, the number of cells at the lower surface membrane which were counted in five randomly selected areas at 400× magnification under an Axioimager M1 fluorescence microscope (Carl Zeiss). The number of invaded cells was counted using ImageJ software. The mean was calculated from the data obtained from each experiment, and each experiment was repeated in three independent runs.
Statistical analysis
All data from at least three independent experiments were normalized to the control. Statistical analysis was performed using GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA). The significance between the two groups was determined using Student’s t test. P<0.01 was considered statistically significant. Error bars represent the mean±standard error of the mean. The half maximal inhibitory concentration (IC50) values and dose response curves were calculated and described using GraphPad Prism 8.0.
RESULTS
Identification of LGZ as a selective PPAR-γ agonist and detecting PPAR-γ expression in BCPAP and K1
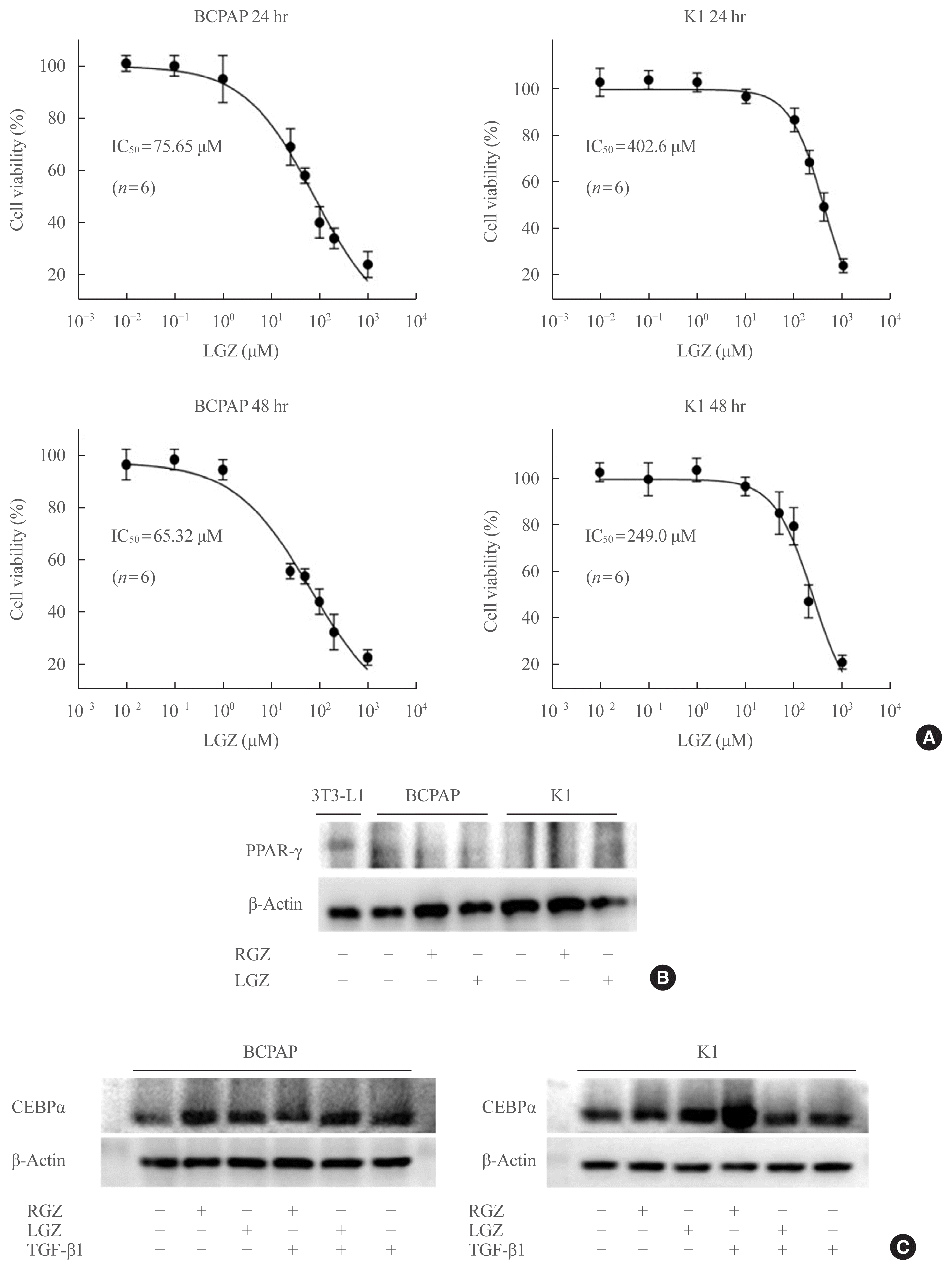
To understand the characteristics of the new PPAR-γ ligand, LGZ, we compared it with the classical TZD, RGZ. The IC50 value of LGZ in PTC has not been previously studied. Through cellular drug toxicity experiments, IC50 value of LGZ in BCPAP cell-line was established as 75.65 μM (24 hours) and 65.32 μM (48 hours), and that in K1 cell-lines was 402.6 μM (24 hours) and 249.0 μM (48 hours), suggesting that BCPAP is more susceptible to LGZ than K1 (Fig. 1A). The PPAR-γ protein expression in BCPAP and K1 was measured using western blotting, and 3T3-L1 cells were used as a positive control (Fig. 1B). BCPAP and K1 cells were treated with RGZ or LGZ for 24 hours. Our results showed that BCPAP and K1 had a very low protein level of PPAR-γ, and no significant difference in PPAR-γ level was observed between RGZ and LGZ treatment compared with the control. As PPAR-γ expression was not definite in BCPAP and K1, expression of C/EBPα, one major protein of the PPAR-γ target genes, was evaluated as a surrogate for PPAR-γ molecular pathway. Western blot result showed that compared with control, C/EBPα expression was increased after RGZ or LGZ treatment, theoretically just as PPAR-γ expression, and this increased expression was attenuated after co-treatment with TGF-β1, but was not attenuated under LGZ co-treatment in BCPAP cells. In K1 cells, on the other hand, C/EBPα expression was increased after RGZ or LGZ treatment (more in LGZ) and decreased after co-treatment with TGF-β1 in LGZ but did not decrease in RGZ, even more increased (Fig. 1C).
TGF-β1 induces epithelial-mesenchymal transition in PTC cells
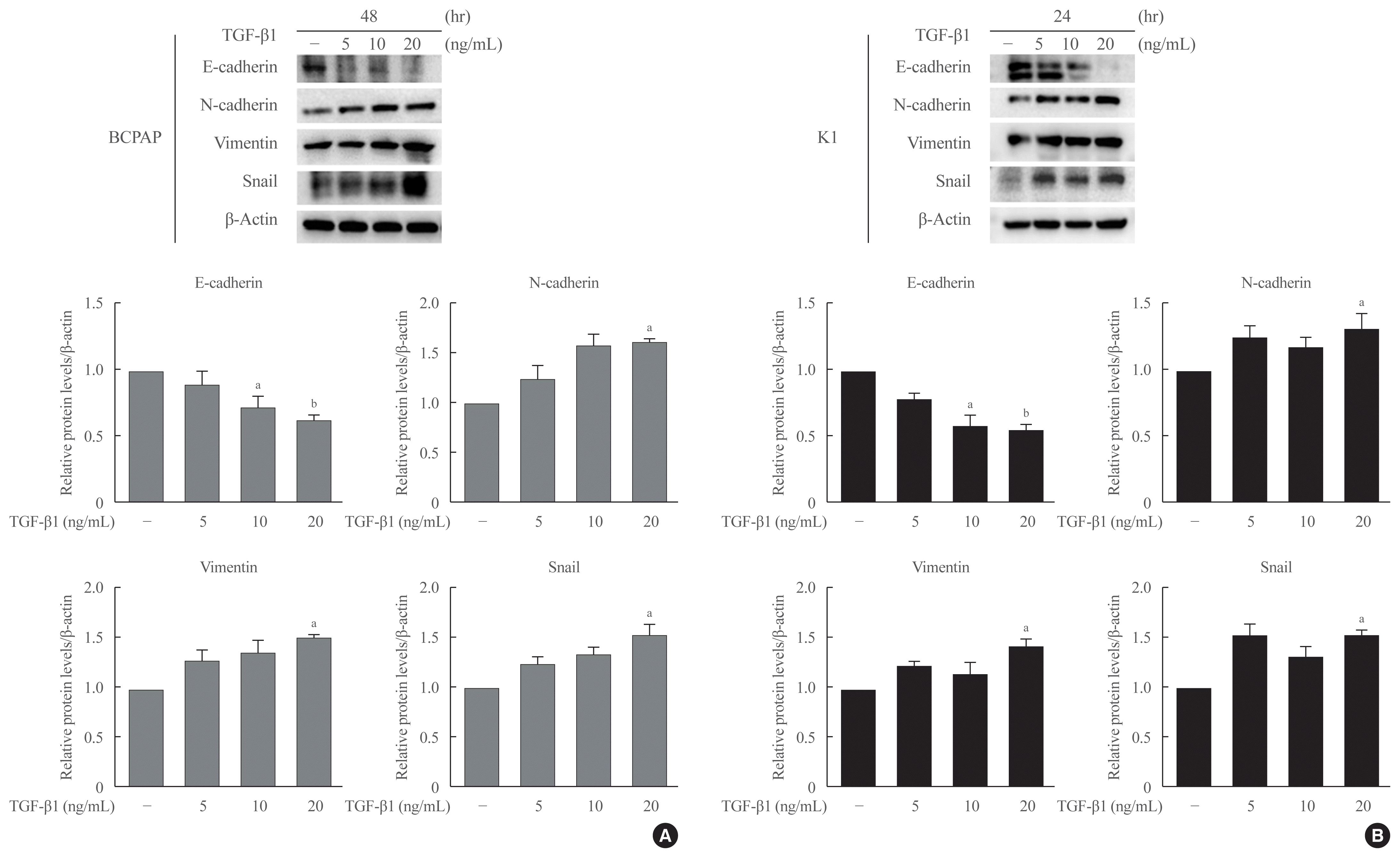
To determine the optimal concentration and time point at which TGF-β1 induces EMT in PTC cells, we used multiple time points (24, 48, and 72 hours) and concentrations (5, 10, and 20 ng/mL) of TGF-β1 to induce EMT in both PTC cells, BCPAP and K1, and evaluated the results using Western blotting. The expression of E-cadherin was significantly decreased, and the expression of N-cadherin, Vimentin, and Snail was increased by the treatment with TGF-β1 at 20 ng/mL for 48 hours in BCPAP and for 24 hours in K1 (Fig. 2).
LGZ and RGZ inhibit TGF-β1-induced EMT in PTC cells
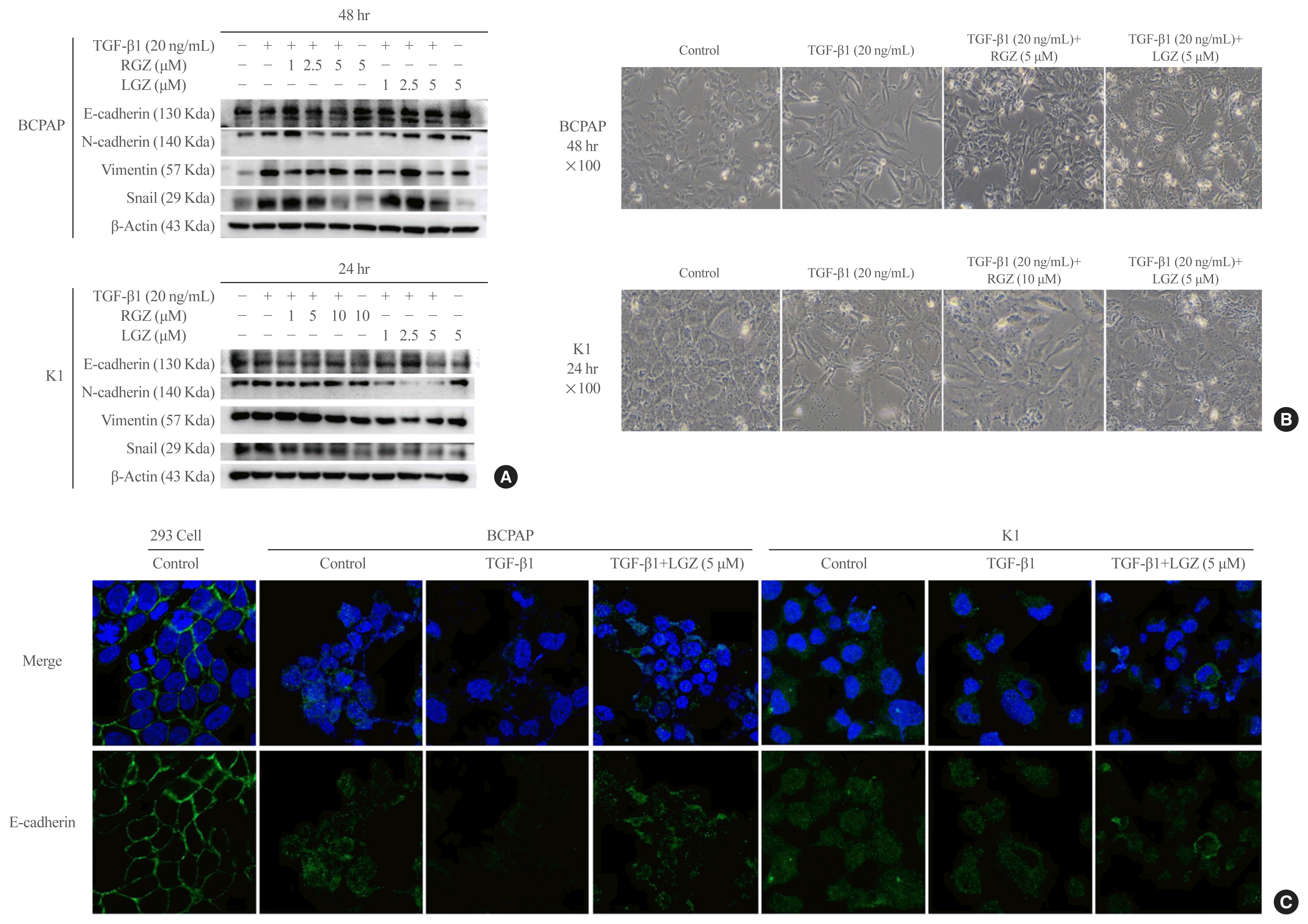
We used western blotting to investigate the effect of PPAR-γ ligands RGZ or LGZ on TGF-β1-induced EMT in PTC cells. The results showed that the expression of E-cadherin was increased and the expression of N-cadherin, Vimentin, and Snail was decreased in a dose-depended manner in response to RGZ and LGZ treatment compared with TGF-β1 treatment alone at 24- and 48-hour time points in PTC cells (Fig. 3A). Stimulation with TGF-β1 (20 ng/mL) in the presence or absence of RGZ (5 μM) or LGZ (5 μM) at 48 hours in BCPAP and RGZ (10 μM) or LGZ (5 μM) at 24 hours in K1 showed that the typical cellular morphological changes corresponding to the EMT phenotype, including loss of cell-to-cell junctions, and increased intercellular space, were induced and reversed by RGZ and LGZ. In contrast, PPAR-γ ligands completely prevented the cells from undergoing the EMT morphological changes induced by TGF-β1 (Fig. 3B). Based on these results, we used 5 μM LGZ (BCPAP and K1) and 5 μM RGZ (BCPAP) and 10 μM RGZ (K1) to maximally inhibit the TGF-β1-induced EMT in the following experiments. Immunocytochemical staining confirmed that treating with LGZ 5μM recovered E-cadherin expression, which was reduced by TGF-β1 in both BCPAP and K1 cells (Fig. 3C).
LGZ and RGZ inhibit TGF-β1-induced EMT in PTC cells through the p38 MAPK signaling pathway
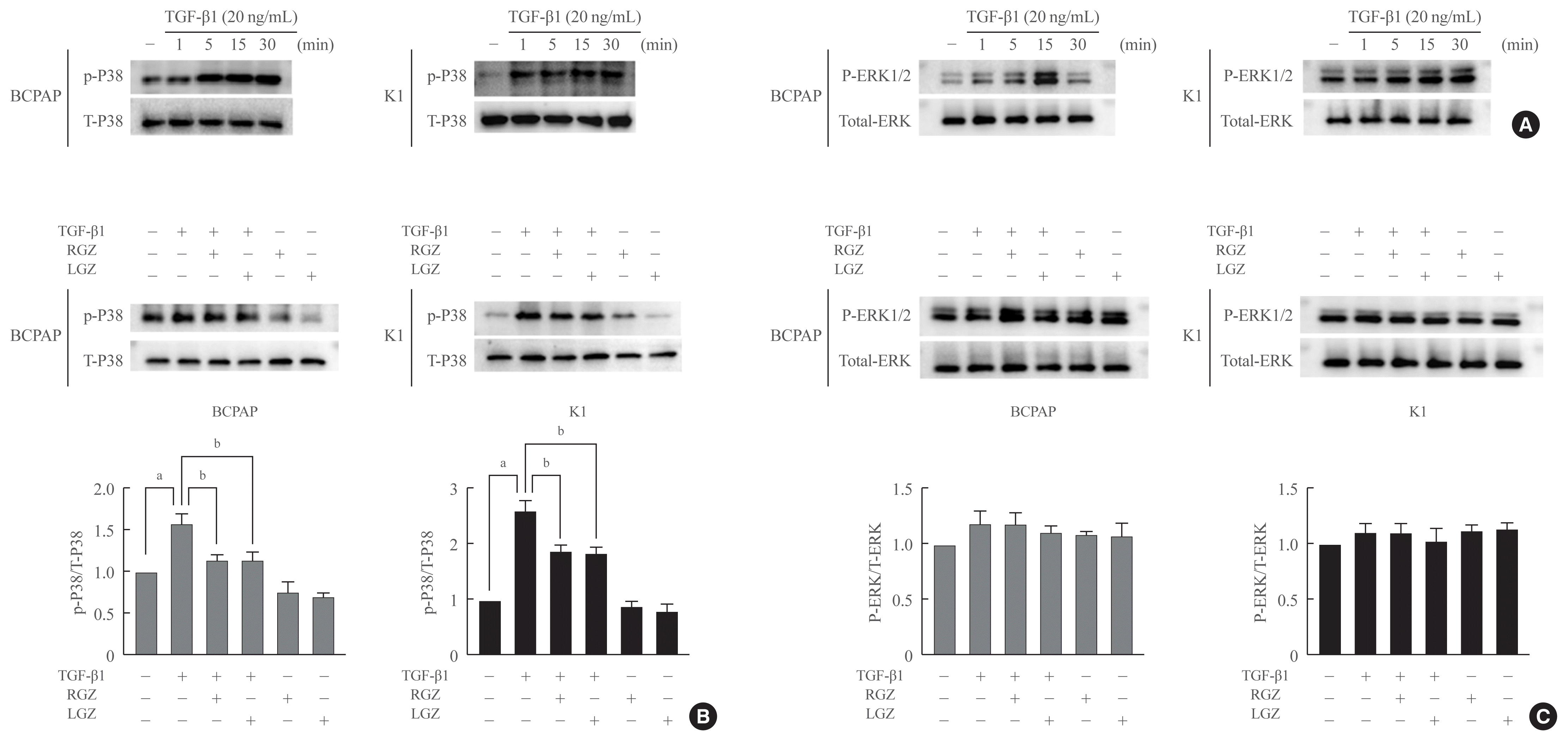
Our experiments showed that p38 MAPK phosphorylation was activated by TGF-β1 in a time-dependent manner. p38 phosphorylation was significantly increased by TGF-β1 in a time-dependent manner (Fig. 4A). RGZ and LGZ decreased the level of p38 MAPK phosphorylation induced by TGF-β1 after 15 minutes. In contrast, total p38 levels were unaffected by the addition of TGF-β1, RGZ, and LGZ (Fig. 4B). We also found that LGZ and RGZ did not have a significant effect on extracellular signal-regulated kinase (ERK) 1/2 phosphorylation induced by TGF-β1 in BCPAP and K1 cells (Fig. 4C).
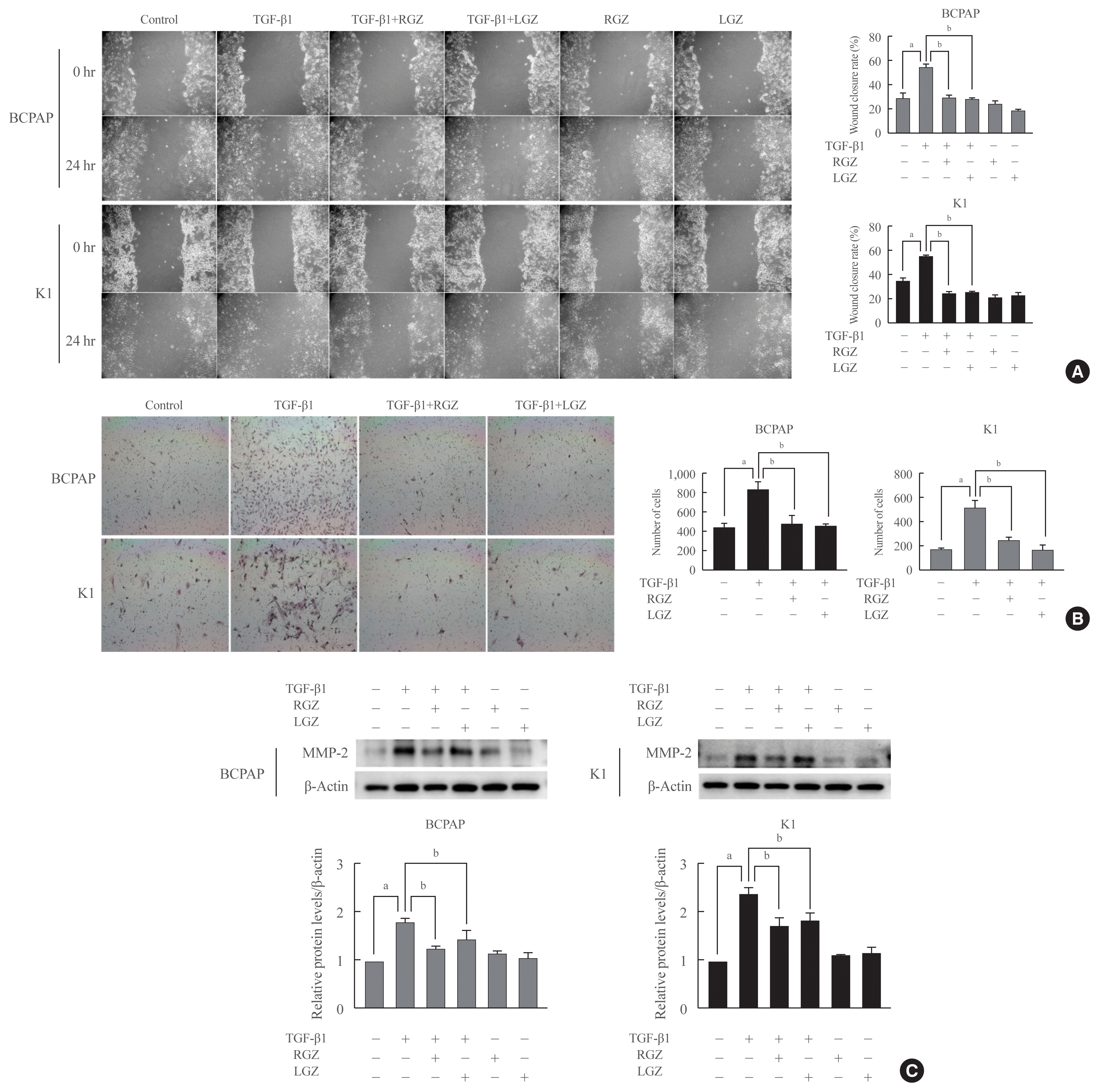
LGZ and RGZ inhibit cell migration, invasion, and MMP-2 protein expression induced by TGF-β1 in PTC cells
To assess the effect of PPAR-γ ligands on TGF-β1-induced PTC migration and invasion, we performed wound healing and Matrigel invasion assays. BCPAP and K1 were pretreated with RGZ and LGZ for 30 min and then treated with TGF-β1 for 24 hours. As the results showed, RGZ and LGZ significantly inhibited TGF-β1-induced cell migration in PTC cells (Fig. 5A). However, RGZ or LGZ stimulation alone did not affect PTC cell migration. In Matrigel invasion, similar to the migration assay results, RGZ and LGZ significantly reduced TGF-β1-induced invasion (Fig. 5B). Furthermore, we assessed MMP-2 expression in PTC cells using western blotting. In both BCPAP and K1 cells, baseline MMP-2 expression was not observed but could be induced by TGF-β1. RGZ and LGZ significantly inhibited TGF-β1-induced MMP-2 expression (Fig. 5C). These results suggest that RGZ and LGZ can inhibit the migration and invasion of thyroid cancer cells in the EMT induced by TGF-β1.
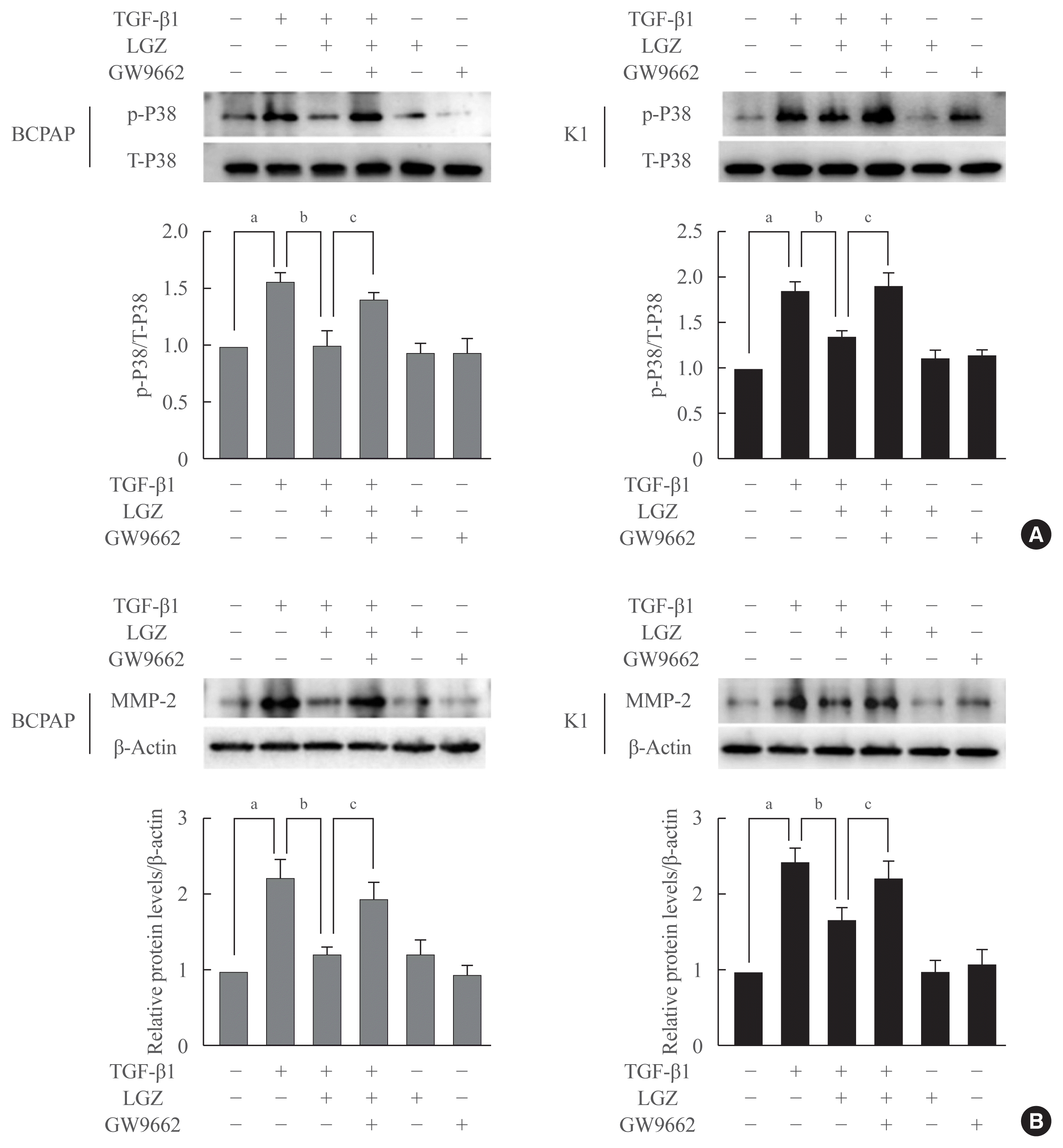
Antitumor effects of LGZ are PPAR-γ dependent
To further assess whether the effects of LGZ on phosphorylation of p38 and MMP-2 expression were PPAR-γ dependent, we used the PPAR-γ selective antagonist GW9662 to block PPAR-γ activity. BCPAP and K1 cells were pretreated with GW9662 for 2 hours, and then stimulated with TGF-β1. GW9662 restored LGZ-induced downregulation of p38 and MMP-2 expression in both BCPAP and K1 cells (Fig. 6), indicating that the effects of LGZ on phosphorylation of p38 and MMP-2 secretion are PPAR-γ-dependent.
DISCUSSION
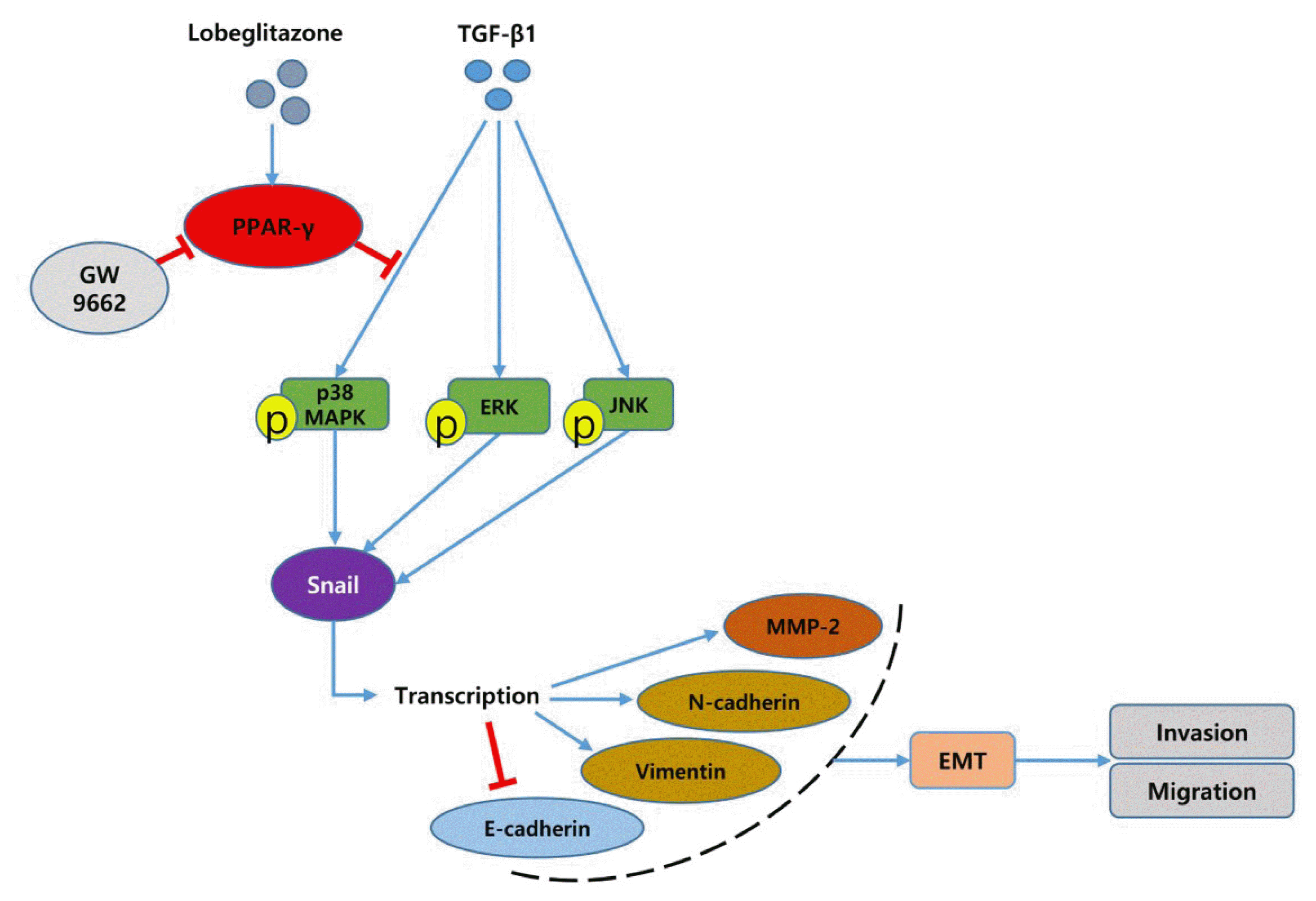
In this study, we investigated the role of LGZ in metastatic behavior of PTC cells. Activation of PPAR-γ by TZDs can inhibit cellular proliferation, migration, and invasion in many types of cancer cells through a PPAR-γ-dependent or -independent pathway [29,30]. These findings indicate that PPAR-γ ligands have the potential to prevent cancer metastasis. In our study, we showed that LGZ and RGZ could prevent TGF-β1-induced EMT, migration, and invasion in PTC cells. We used different time points to treat TGF-β1 (48 hours in BCPAP and 24 hours in K1) because these conditions indicated the typical EMT phenotypes with peak changes of EMT-related molecular markers. Dosages of LGZ (5 μM) and RGZ (10 μM) were in accordance with the reference experiments of EMT in renal cells (LGZ) [31], lung cancer cells and anaplastic thyroid cancer cells [21,25]. Furthermore, LGZ also downregulated MMP-2 expression and p38 phosphorylation induced by TGF-β1 in a PPAR-γ-dependent manner (Fig. 7). Although, PPAR-γ is known as nuclear receptors, when upon ligand binding, PPAR-γ can exert both nuclear genomic and cytoplasmic non-genomic effects. Cytoplasmic PPAR-γ can also modulate MAPKs signaling pathways and it occurs in a receptor-dependent or receptor-independent manner [32,33].
It was shown that when PPAR-γ was overexpressed in BCPAP cells lacking PPAR-γ expression, there was increased growth and increased pRb and cyclin A and B1 levels [34]. However, there was no rigorous correlation between the degree of PPAR-γ expression and functional level and biological response to TZDs, such as RGZ, in thyroid cancer cell lines [25]. Similar to these results, we did not observe increased PPAR-γ protein levels in response to the treatment by LGZ or RGZ in BCPAP and K1. Thus, we chose C/EBPα, one major target gene, as a surrogate of PPAR-γ expression, to evaluate the effects of PPAR-γ agonists and TGF-β1 on PPAR-γ molecular pathway.
Many target genes of PPAR-γ have been already reported, including Fabp4/aP2, C/EBPα, Cfd/Adipsin, CD36 and lipoprotein lipase (LpL) [35–37]. Among them, C/EBPα is a member of subfamily of the basic region leucine zipper transcription factors [38]. It was shown that pioglitazone induced expression of a broad array of PPAR-γ target genes including C/EBPα in mice with paired box gene 8 fusion protein thyroid carcinomas [39]. Previous researches also reported that C/EBPα can inhibit lung cancer development through the p38α MAPK pathway and suppress lung cancer cell invasion and migration [40,41]. As the results showed, C/EBPα expression was increased and decreased to varying degrees by RGZ or LGZ when co-treatment with TGF-β1, maybe due to different mutational profiles of two thyroid cancer cell-lines [42]. Most of all, the relationship between one PPAR-γ target gene, C/EBPα and the EMT-related genes also has been well recognized. Through inhibiting the expression of key mesenchymal markers, C/EBPα prevents EMT-mediated tumorigenesis and maintains epithelial hemostasis [43].
We showed that the potent, selective, and irreversible PPAR-γ antagonist GW9662 prevented LGZ-mediated downstream processes, including the p38 MAPK pathway and MMP2 expression. The transactivation of the different genes by PPAR-γ activation may vary in different cells according to response elements [44].
In earlier studies, preventing the depletion of E-cadherin expression blocked the gain of mesenchymal markers and functional phenotypes of motility and invasion during EMT in human breast cancer and hepatoma cancer cells [45,46]. Activation of PPAR-γ can inhibit the loss of E-cadherin induced by TGF-β1, maintaining a differentiated epithelial phenotype in human prostate cancer cell lines [47]. Therefore, preventing the loss of E-cadherin using TZDs may be another important strategy to inhibit EMT. The present study showed that in an in vitro EMT model induced by TGF-β1 treatment, LGZ and RGZ increased E-cadherin expression and decreased N-cadherin, Vimentin, and Snail expression in PTC cells in a dose-dependent manner. Thus, LGZ has clinical potential as an anti-tumor therapy for PTC metastasis.
Plausible mechanisms underlying the effects of PPAR-γ ligands on TGF-β1 induced EMT, via a Smad or non-Smad pathway, may be cell type-specific and depend on the various types of somatic mutations in cancer cells. Although two PTC cell lines used in our study, BCPAP and K1, have common BRAFV600E mutation, which is the most frequent driver mutation (nearly 80%) in Korea, these two cell types have different mutational profiles [42]: K1 has phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform (PI3KCA) mutation (E542K) in addition to BRAFV600E mutation so may show different response from BCPAP to TGF-β1 and PPAR-γ agonists, because of constitutive kinase activation. Contrary to expectations, however, BCPAP and K1 showed almost similar results, indicating PPAR-γ agonists might be effective to PTC cells without regard to mutational differences.
Although TGFβ/Smad signaling has been recognized as the most important pathway in EMT, the non-Smad pathway, including MAPK signaling pathways, might participate in EMT and tumor progression in thyroid cancer [48]. The execution of the EMT program in response to TGF-β depends on the initiating events during tumor microevolution [49]. In BRAFV600E mutated PTC, constitutive activation of the MAPK pathway promotes tumor progression. P38, the major component of the MAPK pathway in the non-SMAD pathway, is phosphorylated by TGF-β1, which binds to its cell surface receptors (TβRI, TβRII) [50,51]. MAPK can activate avian erythroblastosis virus E26 (v-ets) oncogene homolog-1 (ETS-1) and activated ETS-1 cooperates with Snail 1 to upregulate MMP expression [52]. Consistently, we showed that the MAPK-ERK and p38 phosphorylation as well as the increased expression of Snail and MMP-2 were induced by TGF-β1 in a time-dependent manner. Inhibition of MAPK-ERK activity could block EMT process in normal murine mammary gland epithelia cell [53], and activation of MAPK p38 may lead to EMT by TGF-β1 in mammary epithelial cells [54]. It was also demonstrated that MAPK p38-driven MAPK-activated protein kinase 2 regulated the invasive ability of bladder cancer through modulating MMP-2 and MMP-9 activities [55]. Therefore, the MAPK-ERK or p38 signaling pathway might be essential for the EMT process and tumor metastasis induced by TGF-β1 [56]; and inhibition of MAPK or p38 activation could completely suppress the EMT process.
The effects of RGZ and troglitazone on TGF-β1-induced Smad signaling pathway in lung and pancreatic cancer cells have been well recognized [21]. A previous study demonstrated that pioglitazone had an anti-proliferative effect through the MAPK/AKT cascade as well as the TGFβ/Smad system, thereby exerting anti-metastatic effects by repressing EMT process in human non-small lung cancer cells [57]. Consistent with this observation, both LGZ and RGZ downregulated the MAPK p38 phosphorylation induced by TGF-β1 in our results. Similar to our results, RGZ suppressed fibrotic response by lowering p38 activation but not ERK activation in human pterygium fibroblasts [58]. However, LGZ and RGZ did not suppress the phosphorylation of TGF-β1-induced ERK, another key player in the MAPK pathway. One previous study demonstrated that RGZ inhibited phosphorylation of ERK and Smad2/3 induced by TGF-β1 and no effect on the MAPK p38 in chondrocytes [59]. However, our study suggested that RGZ and LGZ might selectively suppress MAPK p38 to inhibit EMT induced by TGF-β1 in PTC.
In conclusion, the present study revealed that LGZ abrogated EMT, cellular migration, and invasion induced by TGF-β1 in PTC cells through MAPK p38 signaling. Thus, LGZ has a potential to treat a later-stage PTC with metastasis, and we carefully predict new indication of LGZ above anti-diabetic drug. However, as this study did not show any clinical efficacy of LGZ in in vivo model, it should be evaluated whether a PPAR-γ ligand, LGZ may have a clinical role in suppressing metastatic tumors in animal models by aggressive PTC in the near future.
 XML Download
XML Download