

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hypertension is a risk factor for cardiovascular disease, and accelerates the progression of diabetic nephropathy (DN) [1]. The correlation between obesity and hypertension has been well established. Obesity activates the sympathetic nervous system and increases the activity of the renin-angiotensin system (RAS), leading to hypertension [2–4]. In addition, excessive fat accumulation induces hypercholesterolemia, which can damage the kidneys and promote atherosclerosis [5]. Abnormal lipid accumulation in branching vessels thickens the vascular walls and narrows the lumen, leading to increased peripheral resistance [6,7]. Moreover, obesity contributes to increased renal sodium reabsorption due to fat accumulation in the kidneys. Despite increased tubular reabsorption, renal vasodilation and glomerular hyperfiltration are increased to maintain sodium balance [8–10]. In DN, glomerular hyperfiltration is the primary cause of glomerular damage, resulting in albuminuria [11]. Moreover, activation of the intrarenal RAS, increased sympathetic activity, oxidative stress, and increased activity of renal nicotinamide adenine dinucleotide phosphate oxidase IV (NOX4) is considered to be an underlying mechanism of DN [12]. Therefore, preventing hypertension and obesity is one way to prevent DN [13]. A previous study reported improved kidney function in non-diabetic patients with chronic kidney disease (CKD) after treatment with antihypertensive drugs [14]. Interestingly, albuminuria and blood pressure levels in type 2 diabetic patients reduced after 12 weeks of treatment with angiotensin-converting enzyme (ACE) inhibitor [15,16]. These results suggest that lowering blood pressure and RAS activity is a therapeutic strategy for preventing renal failure. Moreover, inhibition of oxidative stress has been shown to attenuate the symptoms of DN, including albuminuria and renal sclerosis, in an experimental diabetic animal model [17,18]. Recently, in patients with CKD, new treatments using combinations of various drugs, such as antihypertensives, RAS inhibitors, and angiotensin II type I receptor blockers, have been investigated [19]. Tetrahydrocurcumin (THU), a major metabolite of curcumin (CUR), exhibits several pharmacological activities, including antihypertensive, antitumor, and antidiabetic properties by reducing lipid peroxide levels, fasting blood glucose levels, and hypertension [20–22]. In an experiment comparing the activities of CUR and THU, it was confirmed that the expression levels of cyclooxygenase 2 and nuclear factor-κB were reduced more effectively by THU treatment than by treatment with CUR [23]. To date, the underlying mechanisms of THU in alleviating kidney injury have not been investigated in an obese or a diabetic animal model. Therefore, the aim of this study was to investigate the protective effect of THU on kidney injury and high systolic blood pressure (SBP) in high-fat diet (HFD)-induced type 2 diabetic mice. An animal model of type 2 diabetes with albuminuria and DN was induced by feeding HFD for 12 weeks [24], and was selected to investigate the protective effect of THU. Here, we hypothesized that THU might have a beneficial effect on kidney injury by lowering high blood glucose levels and preventing an increase in blood pressure.
Go to : 
METHODS
Animal study
Eight-week-old male C57BL/6J mice, weighing 20 to 25 g, were purchased from Daehan Bio Link Co. (Eumseong, Korea). Mice were acclimatized for 7 days under constant room temperature (22°C±2°C), 60%±5% humidity, and a 12-hour light/dark cycle. The mice had free access to water and were fed a regular diet (RD). Thereafter, the mice were randomly divided into four groups (n=10 in each group): RD with vehicle (RD+Veh), HFD with vehicle (HFD+Veh; HFD defined as diet providing 60% of its calories from fat; Research Diets Inc., New Brunswick, NJ, USA), HFD with THU 50 mg/kg/day (HFD+ THU50), and HFD with THU 100 mg/kg/day (HFD+THU100).
THU was intragastrically administered to the HFD-fed mice. Food intake and body weight of each mouse were measured weekly for the duration of the experiment. After 12 weeks, the serum and kidney tissues were collected and stored at −80°C until further processing. All experiments were approved by the Institutional Animal Care and Use Committee of Yonsei University Wonju College of Medicine (YWC-170802-2).
Metabolic parameter measurements
At the end of the experiment, the mice were fasted for 6 hours. Thereafter, the serum was collected. Serum glucose, triglyceride (TG), total cholesterol (TC), aspartate transaminase (AST), and alanine transaminase (ALT) levels were measured using assay kits (ASAN Pharmaceutical, Seoul, Korea). Serum insulin levels were measured using a mouse insulin ELISA kit (Shibayagi, Shibukawa, Japan). The homeostasis model assessment for insulin resistance (HOMA-IR) values were calculated as follows: HOMA-IR=fasting serum glucose (mmol/L)×fasting serum insulin (μU/mL)/22.5.
Glucose and insulin tolerance tests
Blood glucose concentrations were measured by placing a small drop of blood taken from the tip of the tail on a glucose meter strip (Auto-Check, Diatech Korea Co., Seoul, Korea). Fasting blood glucose was measured after 6 hours of daytime fasting. Glucose tolerance test and insulin tolerance test were performed by intraperitoneally injecting glucose (1 g/kg body weight) or insulin (0.75 U/kg body weight; Humulin R, Eli Lilly and Company, Indianapolis, IN, USA) solutions, respectively. Blood glucose concentrations were measured at 0, 15, 30, 60, 90, and 120 minutes.
Renal function test
A 24-hour urine collection was performed for each mouse in a metabolic cage every 4 weeks until the end of the experiment. In the metabolic cages, mice were allowed access to water ad libitum, although no access to food was provided. Renal function tests, evaluating urinary albumin, creatinine concentration, and the albumin-creatinine ratio (ACR) were carried out. Creatinine clearance was performed to estimate the glomerular filtration rate (GFR). Creatinine clearance was calculated as urinary creatinine×urine volume×1,000/serum creatinine/1,440 and was expressed as μL/min. Urine and plasma creatinine concentrations were assessed using a commercial test kit (Exocell, a division of Ethos Biosciences Inc., Newtown Square, PA, USA).
Histological examination of the glomerulus and vascular morphology
Paraffin-embedded kidney tissues were stained with hematoxylin and eosin (H&E) for the examination of the glomeruli and the vascular morphology, and the H&E-stained sections were visualized, for the determination of glomerular volume, vascular wall thickness, cross-sectional area, and nuclear count of the vascular smooth muscle cells, using a microscope equipped with a charge-coupled device camera at 400× magnification (Pulnix, Sunnyvale, CA, USA). Glomerular histopathology and renal fibrosis were also investigated by immunohistochemical (IHC) staining for nephrin, fibronectin, type IV collagen, NOX4, and monocyte chemoattractant protein 1 (MCP-1). Additionally, intrarenal RAS component expression was detected by the IHC staining of ACE and angiotensin 1 receptor (AT1R).
Ultrastructural examination of glomerular changes
The renal cortex was partially excised and immersed in a buffer solution for transmission electron microscopy (TEM). The glomerular basement membrane (GBM) thickness, foot podocyte width, and slit pore density were investigated using TEM at 15,000× magnification.
Western blot analysis
Renal expression of nephrin (1:1,000, sc-377246), fibronectin (1:1,000, sc-8422), type IV collagen (1:1,000, sc-11360), ACE (1:1,000, sc-23909), NOX4 (1:1,000, sc-30141), MCP-1 (1:2,000, sc-28879) (all from Santa Cruz Biotech, Santa Cruz, CA, USA), and AT1R (1:1,000, ab-18801) (Abcam, Cambridge, UK) was detected by Western blot analysis. Renal cortex tissues were homogenized in protein lysis buffer. All samples were centrifuged at 13,000 ×g for 20 minutes. The supernatants were mixed with the sample buffer and heated for 5 minutes at 95°C. Sample aliquots containing 15 to 30 μg of protein were loaded on sodium dodecyl sulphate polyacrylamide gel electrophoresis (8% to 12%) and subsequently transferred to polyvinylidene difluoride membranes. The membranes were incubated at 4°C overnight with primary antibodies and washed three times in Tris-buffered saline with 0.1% Tween 20. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. The gels were visualized using an enhanced chemiluminescence gel electrophoresis system (Amersham Biosciences, Buckinghamshire, UK). The intensity of the bands was measured using ImageJ software version 1.50i (NIH, Bethesda, MD, USA).
Measurement of systolic blood pressure
SBP was monitored at the start of the experiment and after every 2 weeks for the duration of the experiment using mouse tail-cuff photoplethysmography (BP-2000 blood pressure analysis system, Visitech Systems Inc., Apex, NC, USA). The mice were placed in a restrainer at 34°C for 5 minutes. Eight consecutive SBP values were recorded. Furthermore, heart rate was obtained from the data transformation of arterial pulses.
Statistical analysis
All data are presented as the mean±standard error of the mean. Statistical analysis included one-way analysis of variance (ANOVA) and Tukey’s post hoc test for multiple comparisons, and was conducted using SPSS Statistics software version 20.0 (IBM Corp., Armonk, NY, USA). Statistical significance was set at P<0.05.
Go to :
RESULTS
Tetrahydrocurcumin prevents increased body weight and food intake in HFD-fed mice
HFD-fed mice showed a gradual increase in body weight (Fig. 1A) and food intake (Fig. 1B) throughout the experimental period. Significant differences in body weight and food intake were observed after 4 and 5 weeks, respectively (P<0.05 vs. RD-fed mice). Treatment with THU, particularly at the dose of 100 mg/kg/day, prevented the increased body weight and food intake (P<0.05 vs. HFD+Veh) (Fig. 1).
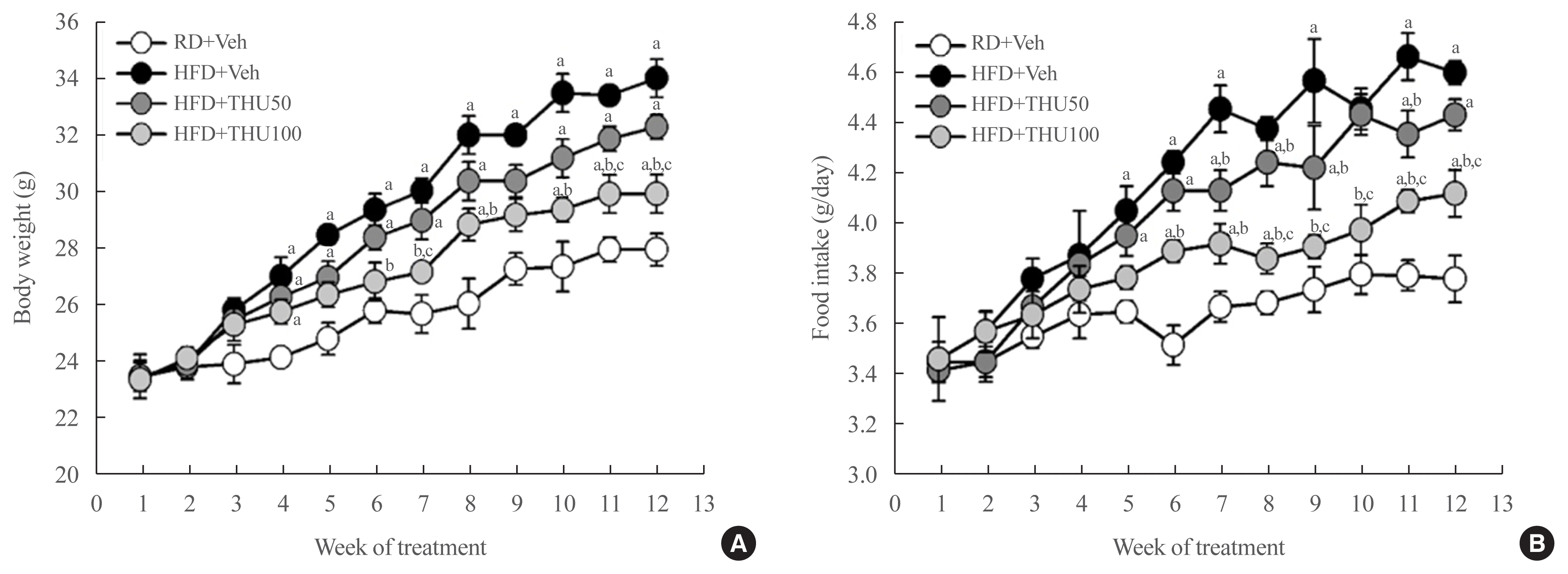 | Fig. 1Tetrahydrocurcumin (THU) prevents an increase in body weight and food intake in high-fat diet (HFD)-fed mice. C57BL/6J mice were fed a regular diet (RD) or HFD for 12 weeks, and THU (50 or 100 mg/kg/day) was intragastrically administered with HFD. Changes in body weight (A) and food intake (B) during the experimental period. aP<0.05 vs. RD+vehicle (Veh); bP<0.05 vs. HFD+Veh (n=8 to 10 per group); cP<0.05 vs. HFD+THU50.
|
Tetrahydrocurcumin alleviates glucose intolerance, insulin resistance, and renal dysfunction in HFD-fed mice
HFD-fed mice exhibited hyperglycemia, insulin resistance, and dyslipidemia (Table 1). THU effectively reduced serum glucose, insulin, TC, TG, AST, and ALT levels in these mice in a dose-dependent manner (Table 1). Moreover, THU potentially attenuated glucose intolerance (Fig. 2A, B) and insulin resistance (Fig. 2C, D) in HFD-fed mice, which is an important observation, as the alteration of metabolic parameters and glucose homeostasis plays a dominant role in the pathogenesis of kidney dysfunction. The present study demonstrated that consumption of HFD increased ACR, kidney weight, and GFR (Fig. 2E–G). Interestingly, treatment with THU, especially at a high dose (100 mg/kg/day), significantly decreased these defects (P<0.05). These data suggest that THU has beneficial effects on glucose intolerance, insulin resistance, and kidney function.
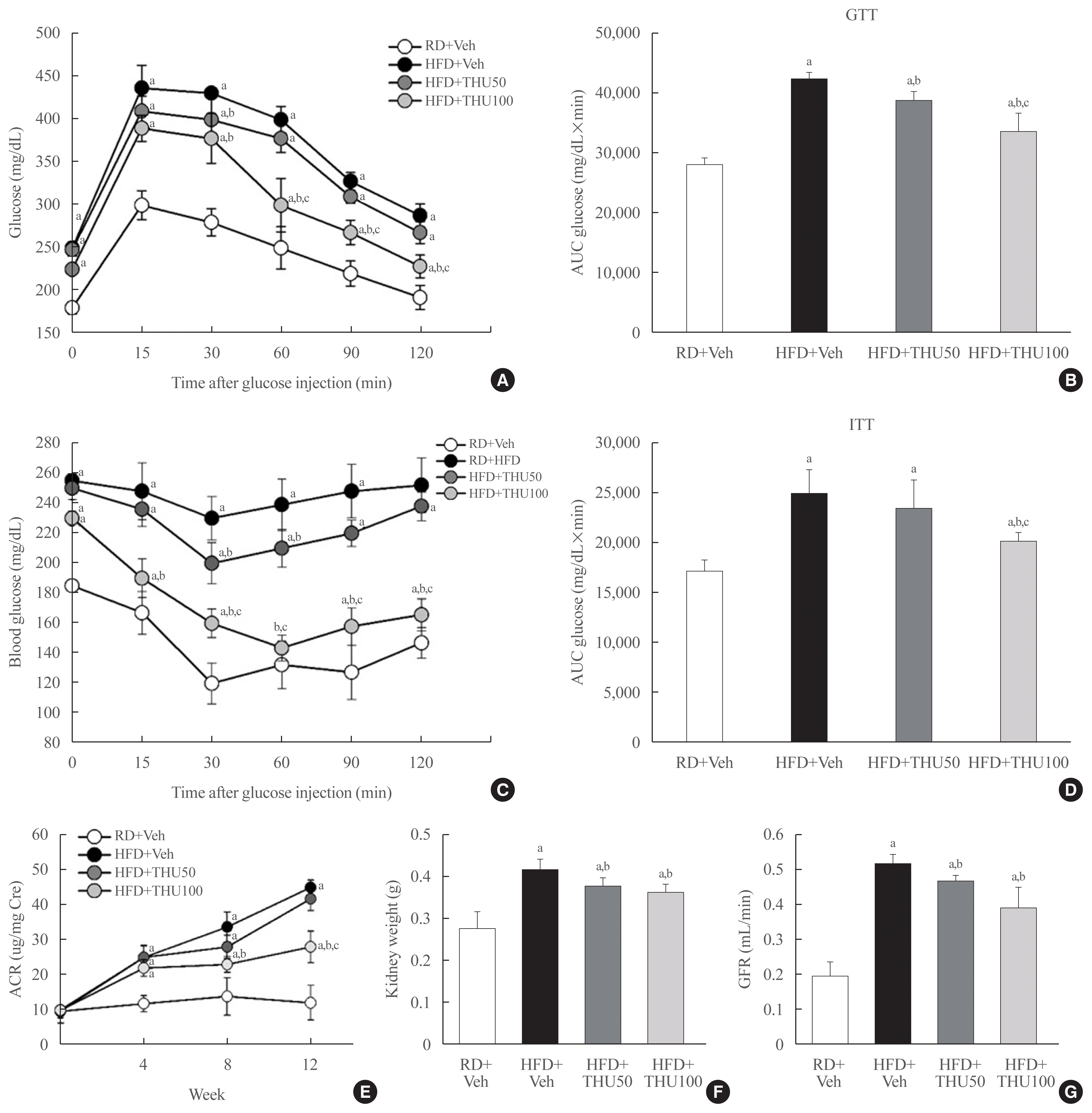 | Fig. 2Tetrahydrocurcumin (THU) alleviates glucose intolerance, insulin resistance, and renal dysfunction in high-fat diet (HFD)-fed mice. Intraperitoneal injection (IP) glucose tolerance test (GTT) and IP insulin tolerance test (ITT) (A, C) at 10 weeks. Levels of area under curve (AUC) in GTT and ITT (B, D). Kidney weight differences among the four groups (E). Albumin-creatinine ratio (ACR) analysis in urine collected at 24 hours (F). Changes in the glomerular filtration rate (GFR) after THU administration (G). RD, regular diet; Veh, vehicle. aP<0.05 vs. RD+Veh; bP<0.05 vs. HFD+Veh; cP<0.05 vs. HFD+THU50 (n=8 to 10 per group).
|
Table 1
Tetrahydrocurcumin Improves Impaired Metabolic Parameters in High-Fat Diet-Fed Mice
| Parameter | RD+Veh | HFD | ||
|---|---|---|---|---|
| Veh | THU50 | THU100 | ||
| Serum glucose, mg/dL | 189.78±10.99 | 289.08±14.23a | 276.47±15.30a,b | 254.42±11.93a,b,c |
| Serum insulin, pg/mL | 99.80±8.09 | 234.31±15.39a | 202.02±22.21a,b | 146.58±15.14a,b,c |
| HOMA-IR index | 1.35±0.21 | 5.07±0.32a | 3.90±0.08a,b | 2.56±0.15a,b,c |
| Serum total cholesterol, mg/dL | 167.58±19.25 | 323.34±20.65a | 295.12±15.39a,b | 259.10±11.37a,b,c |
| Serum triglycerides, mg/dL | 80.25±9.25 | 151.47±21.39a | 147.64±18.32a | 119.54±15.02a,b,c |
| Serum AST, unit/mL | 89.34±10.58 | 132.76±12.36a | 128.09±11.25a | 109.33±9.20a,b,c |
| Serum ALT, unit/mL | 40.39±3.25 | 56.08±4.59a | 45.78±6.85a,b | 43.89±7.51a,b |
![]()
Tetrahydrocurcumin prevents glomerular hypertrophy in HFD-fed mice
HFD-induced DN is closely associated with glomerular damage. Histopathological analysis showed glomerular expansion in the HFD-fed mice. However, THU at 100 mg/kg/day reduced glomerular area and volume (Fig. 3A–D). GBM thickness was followed by mesangial expansion, which is a hallmark of DN. HFD-fed mice exhibited increased GBM thickness, and decreased number of slit pores. However, THU treatment reversed the HFD-induced changes in GBM thickness, slit pore numbers, and the foot width of podocytes, particularly in the THU100 group (Fig. 3E–G). These data suggest that treatment with THU at 100 mg/kg/day prevented glomerular hypertrophy and the mesangial expansion induced by HFD consumption.
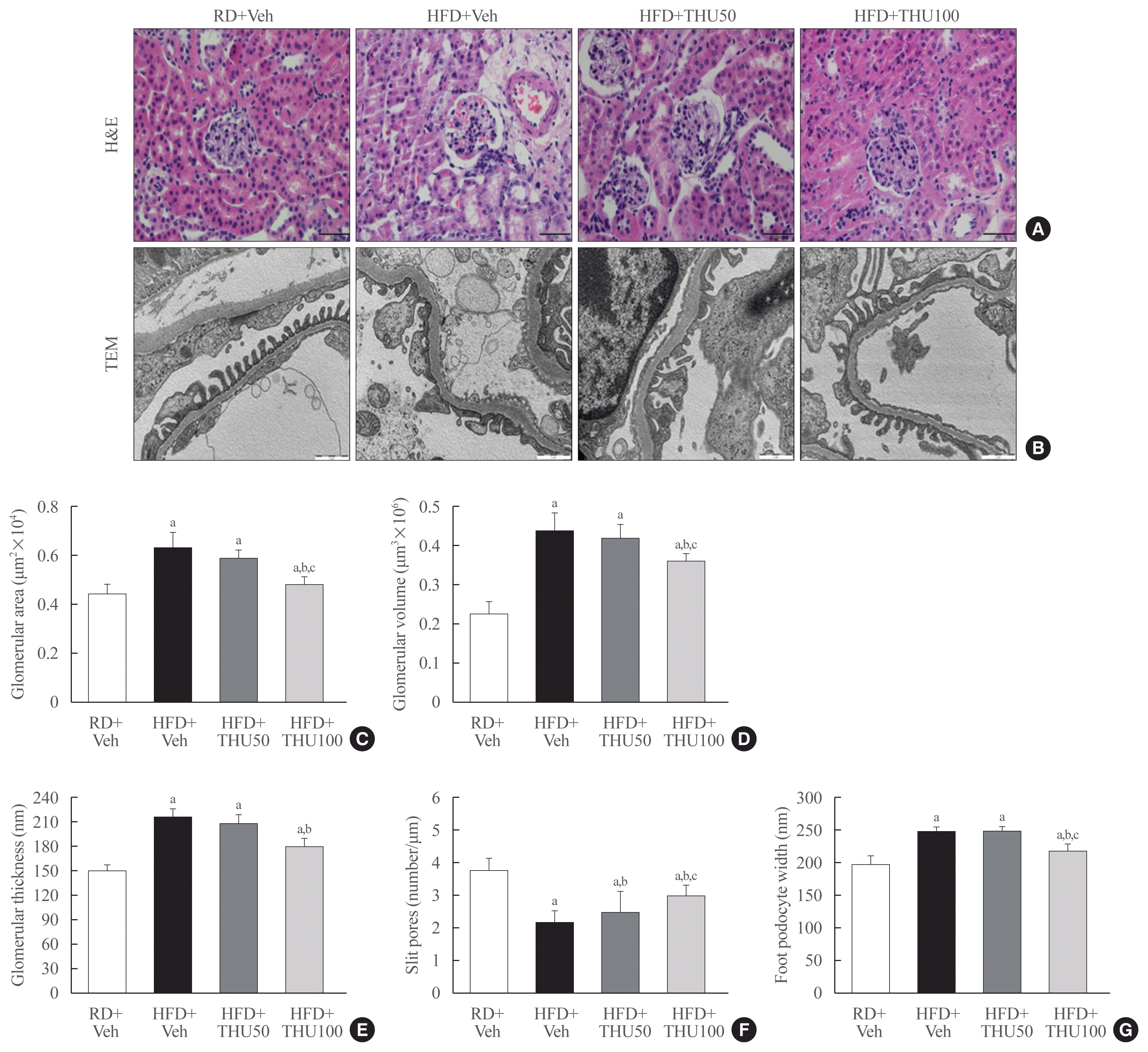 | Fig. 3Tetrahydrocurcumin (THU) protects against glomerular hypertrophy in high-fat diet (HFD)-fed mice. Effect of THU on the enlarged glomeruli of HFD-fed mice. Hematoxylin and eosin staining of the kidneys of all experimental groups (A). Changes in glomerular area and glomerular volume by THU (C, D) (n=8 to 10 per group). Renal ultrastructure changes caused by THU in HFD (B). Quantitative transmission electron microscopy (TEM) data. The glomerular basement membrane (GBM) thickness (E), slit pore numbers (F), and foot process width of podocytes (G) were analyzed by TEM at 15,000× magnification (n=5 per group). RD, regular diet; Veh, vehicle. aP<0.05 vs. RD+Veh; bP<0.05 vs. HFD+Veh; cP<0.05 vs. HFD+THU50 (scale bar=50 μm).
|
Tetrahydrocurcumin restored nephrin expression and decreased extracellular matrix accumulation in the diabetic glomeruli of HFD-fed mice
Nephrin is a key molecule that forms the glomerular barrier. Nephrin levels were decreased in the HFD group. Treatment with THU (100 mg/kg/day) in HFD-fed mice increased the nephrin levels. It also decreased the expression of fibronectin and collagen IV as indicated by IHC staining (Fig. 4A–D), and Western blot assays (Fig. 4E, F). This suggests that THU at a dose of 100 mg/kg/day prevents the accumulation of extracellular matrix (ECM).
 | Fig. 4Tetrahydrocurcumin (THU) restores nephrin expression and decreases extracellular matrix accumulation in the diabetic glomeruli of high-fat diet (HFD)-fed mice. Renal nephrin (A), fibronectin (B), and collagen type IV (C) were subjected to immunohistochemical (IHC) staining. Panel (D) indicates the percentage of positively stained area of each protein in glomeruli. Original magnification was 400× (scale bar=50 μm). Nephrin, fibronectin, and type IV collagen expression were analyzed by Western blotting (E, F). All protein expression analyses using Western blotting were performed on samples of the renal cortex (n=8 to 10 per group). RD, regular diet; Veh, vehicle. aP<0.05 vs. RD+Veh; bP<0.05 vs. HFD+Veh; cP<0.05 vs. HFD+THU50.
|
Tetrahydrocurcumin downregulates ACE-AT1R-NOX4-MCP-1 expression in the glomeruli of HFD-fed mice
The mechanisms of hypertension, oxidative stress, and renal inflammation in HFD-fed mice include the activation of intrarenal RAS, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, and MCP-1. In the glomeruli of the HFD-fed mice, an increase in the stained areas denoting ACE and AT1R expression was observed. Further, expression of NADPH oxidase 4 and of inflammatory signals, such as MCP-1, was also increased, as indicated by IHC staining (Fig. 5A–D). However, the administration of THU100 resulted in a decrease in the positively stained areas corresponding with the aforementioned markers (Fig. 5E). The protein expression levels of ACE, AT1R, NOX4, and MCP-1 in the kidney were consistent with the IHC staining data (Fig. 5F). These results suggest that THU at a dose of 100 mg/kg/day reduces the activity of RAS, and reactive oxygen species (ROS), thus attenuating renal inflammation.
 | Fig. 5Tetrahydrocurcumin (THU) downregulates the expression of angiotensin-converting enzyme (ACE), angiotensin 1 receptor (AT1R), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4), and chemokine C-C motif ligand 2 (MCP-1) in the glomeruli of high-fat diet (HFD)-fed mice. (A, B, C, D) ACE, AT1R, NADPH oxidase 4, and MCP-1 expression indicated by immunohistochemical staining. (E) Panel indicates the positively stained area of each protein in the glomeruli (n=4 to 5 per group; scale bar=50 μm). (F) Western blot analysis of the renal cortex. RD, regular diet; Veh, vehicle. aP<0.05 vs. RD+Veh; bP<0.05 vs. HFD+Veh; cP<0.05 vs. HFD+THU50.
|
Tetrahydrocurcumin attenuates high systolic blood pressure and the remodeling of vascular structures in HFD-fed mice
SBP was elevated, and vascular structures were altered by the long-term administration of HFD. HFD increased the number of nuclei on the tunica surface, the wall thickness, and the cross-sectional area of the blood vessels (Fig. 6A–D). Administration of THU (100 mg/kg/day) reduced the SBP (Fig. 6E). However, the heart rate did not change in any of the groups (Fig. 6F). These data suggest that THU100 prevents vascular remodeling, thereby preventing an increase in SBP.
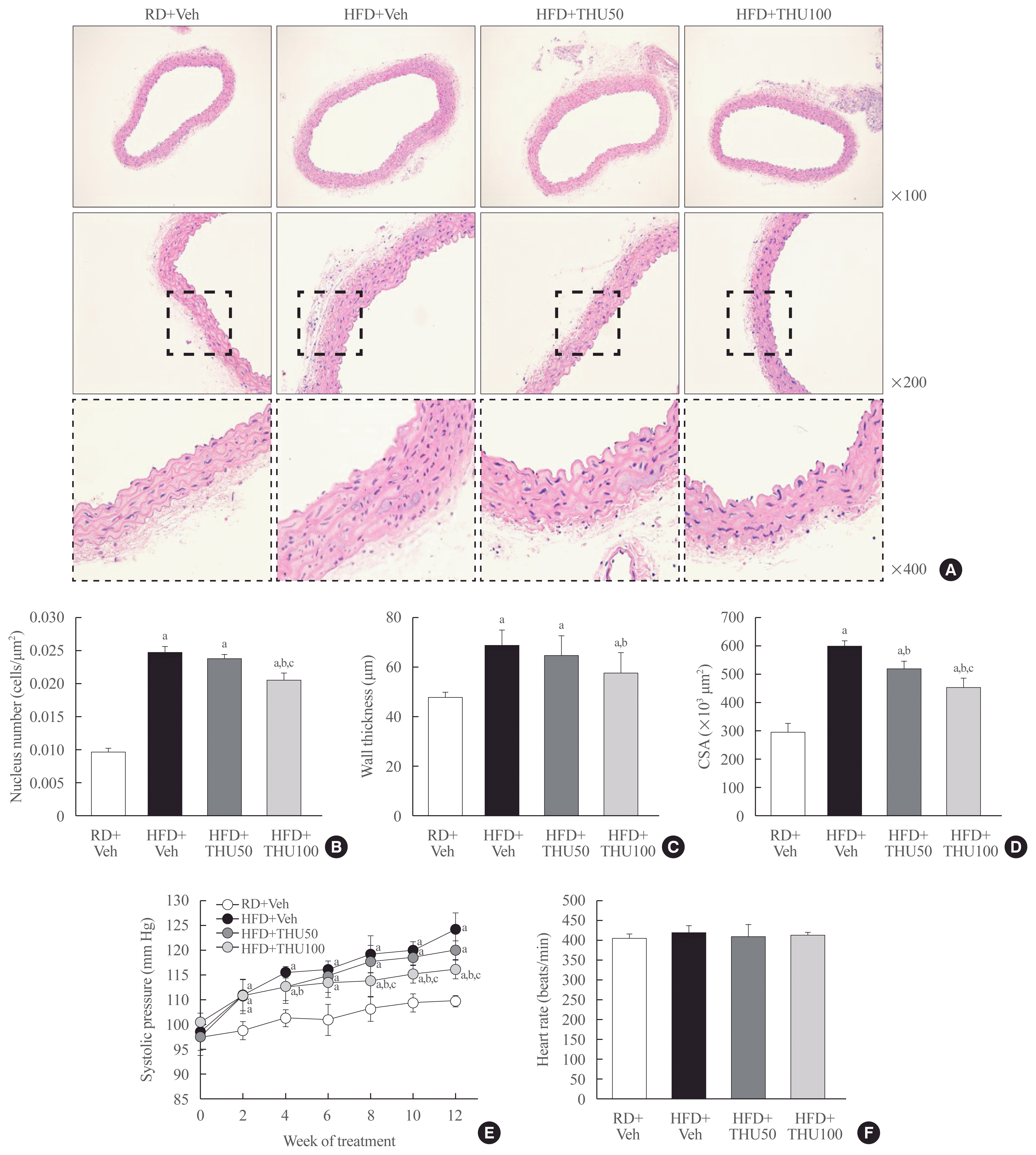 | Fig. 6Tetrahydrocurcumin (THU) attenuates high systolic blood pressure and remodeling of vascular structures in high-fat diet (HFD)-fed mice. Vascular structural changes (A), nucleus count by tunica media area (μm2), vascular wall thickening (μm), vascular cross-sectional area (CSA) (×103 μm2) (B, C, D), systolic blood pressure (E), and heart rate throughout the 12 weeks of treatment (F) (n=8 to 10 per group). RD, regular diet; Veh, vehicle. aP<0.05 vs. RD+Veh; bP<0.05 vs. HFD+Veh; cP<0.05 vs. HFD+THU50.
|
Go to :
DISCUSSION
The present study showed that treatment with THU at a dose of 100 mg/kg/day effectively prevented kidney injury and high SBP in HFD-induced type 2 diabetic mice. In this present study, the administration of an HFD for 12 weeks caused type 2 diabetes in mice, as evidenced by the observation of weight gain, impaired glucose regulation, and insulin resistance, and of associated metabolic disorders, including hyperinsulinemia and dyslipidemia. Albuminuria, glomerular hypertrophy, thickening of the GBM, and podocyte foot detachment were also noted in HFD-fed mice, suggesting that the kidneys were damaged and enriched in ECM components such as collagen and fibronectin. Importantly, HFD consumption increased SBP and induced vascular remodeling. These findings are consistent with a previous study reporting that long-term feeding of HFD for 12 weeks induced obesity, glomerular damage, and vascular structural changes [25].
The occurrence of albuminuria, hypertension, and histopathological changes in the glomeruli of diabetes patients are characteristics of DN, which is a progressive kidney disease. A damaged kidney loses its physiological functions, including that of filtration and reabsorption [26,27], resulting in leakage of albumin into the urine and the retention of waste products in the body.
HFD consumption may exert a deleterious effect on the kidneys by promoting renal inflammation. HFD increases the infiltration of macrophages in adipose tissue and kidneys, leading to an inflammatory response and tissue damage. In addition, HFD may induce kidney injury by promoting glomerular hypertrophy, the thickening of the GBM, and glomerular sclerosis, thereby disrupting renal hemodynamics. Our results are consistent with those of a previous study showing that renal vasodilation, increased renal blood flow, and glomerular hyperfiltration occurred after weight gain [28].
Hyperglycemia is a consequence of HFD-induced type 2 diabetes. It plays an important role in the pathogenesis of kidney injury. Oxidative stress is a well-known pathogenic mechanism of hyperglycemia, which triggers diabetic complications. Oxidative stress triggers the accumulation of angiotensinogen, subsequently activating the RAS [29]. A previous study revealed that all RAS components (ACE, angiotensin II, and AT1R) are extensively expressed in the renal endothelial cells of diabetic rodents [30]. Moreover, ACE activity levels increase along with blood glucose and hemoglobin A1c levels in diabetes patients with nephropathy [31]. These studies suggest that hyperglycemia activates the RAS. The binding of angiotensin II to AT1R activates NADPH oxidase (NOX), which transfers an electron from NADPH to O2, producing the superoxide anion (O2−) [32]. Excess O2− generation causes oxidative stress and oxidative damage to renal tissues. Moreover, NOX-derived ROS were found to be involved in angiotensin II-induced MCP-1 expression in mesangial cells [33].
We demonstrated that THU alleviates kidney injury by suppressing the RAS/NOX4/MCP-1 axis. The possible mechanism of action of THU might involve its antioxidant property and blood glucose-lowering effect. The structure of THU includes a phenol and a β-diketone functional group, which is a common structural feature found in antioxidative molecules [34]. Previous studies have demonstrated that the administration of THU mitigates oxidative stress by scavenging O2− [35]. Thus, the subsequent reduction in the excessive levels of ROS (under the hyperglycemic conditions) potentially suppresses the activation of the RAS/NOX4/MCP-1 axis.
Apart from hyperglycemia-mediated ROS that enhances RAS activation, hyperglycemia directly activates the RAS [30]. The present study showed the beneficial effects of THU for improving insulin sensitivity, and thereby lowering serum glucose levels. The effect of THU on the reduction of hyperglycemia may be partially explained by the antioxidant properties of THU. Attenuation of oxidative stress is directly associated with increased insulin sensitivity by reducing the oxidation of insulin receptors and intracellular glucose transporters [36]. This suggests that THU may prevent the alteration of insulin receptor signaling. Our findings are consistent with those of previous studies showing improved insulin sensitivity in diabetic mice after THU treatment [37,38]. Therefore, attenuation of hyperglycemia alters the activation of the RAS/NOX4/MCP-1 axis under diabetic conditions.
Administration of HFD for 12 weeks induced glomerular hypertrophy and podocyte injury. Treatment with THU at 100 mg/kg/day in HFD-fed mice reduced the foot process width of podocytes, determined morphometrically by electron microscopy, suggesting that THU potentially alleviates podocyte foot process effacement. Podocyte dysfunction is associated with altered insulin signaling pathways [39]. Moreover, treatment with ACE inhibitors or angiotensin II type 1-receptor blockers in patients with DN can reduce podocyte injury [40]. This confirms that the activation of the RAS is implicated in podocyte injury. Furthermore, oxidative stress plays a major role in podocyte injury by enhancing endoplasmic reticulum stress, which may trigger autophagy [41]. The mechanism by which THU alleviates alterations in podocyte function may be associated with the antioxidant properties of THU.
Insulin sensitivity is important for normal podocyte function and glucose regulation. In this study, HFD-fed mice showed insulin resistance and increased HOMA-IR and hyperglycemia; however, this effect was attenuated by treatment with THU does dependently. Oxidative stress-associated obesity and increased inflammation potentially alter insulin action by worsening the insulin receptor interaction and signal transduction. This phenomenon is a leading cause of insulin resistance and hyperinsulinemia. Attenuation of oxidative stress by THU prevents the alteration of insulin receptor signaling, thereby improving insulin sensitivity [36].
THU attenuates high SBP in HFD-fed mice by reducing aortic wall thickness. This effect may be related to the antioxidant properties of THU. The reduction in oxidative stress increases the bioavailability of nitric oxide, a potent vasodilator, leading to increased vascular relaxation and a reduction in SBP.
The cumulative data showed that THU can serve an effective bioactive compound for the attenuation of DN due to its ability to reduce metabolic abnormalities, kidney injury, and the high SBP caused by HFD consumption.
In conclusion, THU exerts a renoprotective effect in HFD-fed mice by inhibiting the intrarenal RAS/NOX4/MCP-1 axis and attenuating insulin resistance and reducing the high SBP.
Go to :
 XML Download
XML Download