

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Sarcopenia is a progressive skeletal muscle disorder characterized by the accelerated loss of muscle mass and function [1,2]. This condition has recently drawn particular attention due to the increased likelihood of negative health outcomes, such as frailty, falls, physical weakness, poor quality of life, and mortality [3,4], and has been officially classified as a disease, reflecting its clinical significance [5]. Although the term “sarcopenia” was first used to describe age-related muscular decline [6], increasing knowledge about muscle metabolism has revealed that sarcopenia could be attributable to heterogeneous causes, including systemic inflammatory diseases, malignancy, organ failure, or medications [2,3,7]. This secondary sarcopenia involving specific causes other than aging is particularly important from a clinical perspective because of its potential reversibility. Therefore, it is critically important to continuously pursue efforts to identify the modifiable causes of sarcopenia in order to extend the healthy lifespan in old age and to reduce the associated unnecessary economic burden on national healthcare systems.
Primary aldosteronism (PA) refers to inappropriate and autonomous hypersecretion of aldosterone from the adrenal glands [8,9]. It is regarded as a good human model to elucidate the role of excess aldosterone in the pathogenesis of diverse diseases. PA accounts for approximately 10% of hypertensive patients and is known to contribute to end-organ damage, especially in kidney, heart, and carotid artery [10,11]. Accumulating evidence now indicates that aldosterone can deleteriously affect muscle phenotypes as well. An animal study showed that the infusion of aldosterone-induced skeletal muscle apoptosis in rats [12]. Furthermore, compared with subjects with nonfunctional adrenal incidentaloma, those with PA had low skeletal muscle mass, leading to an increased risk of sarcopenia [13]. However, it is still unclear whether the role of aldosterone in muscle metabolism is direct or mediated indirectly by factors, such as electrolyte imbalance or impaired glucose uptake [9,14–16]. As one approach to clarify this issue, we investigated the direct effect of aldosterone on in vitro myogenesis and the potential mechanism explaining it.
METHODS
Cell culture and reagents
Mouse C2C12 myoblasts were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 15% fetal bovine serum (FBS), 20 mM hydroxyethyl piperazine ethane sulfonic acid (HEPES), 2 mM L-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Life Technologies Corp., Carlsbad, CA, USA) at 37°C in a humidified atmosphere containing 5% CO2. To induce myogenesis, cells were grown to 90% confluency in maintenance media and switched to differentiation media (DMEM with 2% horse serum) for 3 or 4 days of culture. Recombinant aldosterone (A9477), eplerenone (E6657), and N-acetyl-L-cysteine (NAC, A9165) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Immunofluorescence
Differentiated C2C12 cells were fixed in 4% paraformaldehyde (PFA) for 15 minutes, washed twice with phosphate-buffered saline (PBS), permeabilized in 0.01 M sodium citrate buffer containing 0.1% Triton X-100 for 10 minutes, and washed twice with PBS. The cells were blocked with 2% bovine serum albumin (BSA) in PBS for 1 hour and subsequently incubated with anti-myosin heavy chain (MyHC) antibody (MF20, Developmental Studies Hybridoma Bank, Iowa City, IA, USA) at 4°C overnight. The cells were incubated with Alexa Fluor 555-conjugated secondary antibodies (1:1,000 dilution, Cell Signaling, Danvers, MA, USA) for 1 hour and washed with 0.2% Tween-20 in PBS. Thereafter, they were incubated with 4,6-diamidino-2-phenyindole (DAPI, 1:10,000 dilution, Sigma-Aldrich) for 2 minutes and washed with PBS. The samples were mounted using Fluoromount G (Southern Biotech, Birmingham, AL, USA) and images were captured using a fluorescence microscope (Carl Zeiss, Jena, Germany). The area of the MyHC-stained myotubes was calculated using the ZEN 2 (blue edition) software (Carl Zeiss). Fusion index (%) was calculated using the following equation: 100×number of nuclei in MyHC+ myotubes per total number of nuclei in MyHC+ myocytes and myotubes [17].
Quantitative reverse-transcription polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), in accordance with the manufacturer’s protocol. After first-strand cDNA synthesis with the Superscript III First-Strand Synthesis System (Invitrogen) using oligo dT primers, quantitative reverse-transcription polymerase chain reaction was performed in triplicate on a Light Cycler 480 SYBR Green I Master (Roche, Mannheim, Germany) [18]. The primers for myogenin (NM_031189.2), myocyte enhancer factor 2C (Mef2C; NM_001170537.1), and MyHC (NM_001013397.2) were obtained from Applied Biosystems (Foster City, CA, USA). The threshold cycle (Ct) value for each gene was normalized to the Ct value of 18S rRNA (NR_003278.3).
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1 mM ethylene glycol tetra acetic acid, 1 mM ethylenediaminetetraacetic acid, 0.1% sodium dodecyl sulfate [SDS], 1% sodium deoxycholate, 1 mM Na3VO4, 1 mM sodium fluoride, 1 mM phenyl methyl sulfonyl fluoride, and protease inhibitor cocktail). After 30-minute incubation on ice, the lysates were centrifuged at 14,000 rpm for 20 minutes at 4°C. The protein concentration was measured with a bicinchoninic acid (BCA) protein assay kit (Pierce Chemical Co., Rockford, IL, USA). Protein samples were separated using sodium dodecyl sulfate-polyacrylamide electrophoresis, transferred to polyvinylidene fluoride membranes, and immunoblotted using the following antibodies [19]: MyHC (MF20), myogenin (sc-12732, Santa Cruz Biotechnology, Dallas, TX, USA), mineralocorticoid receptor (MR) (ab64457, Abcam, Cambridge, MA, USA), and β-tubulin (T2200, Sigma-Aldrich).
Viability assay
Cell viability was measured using cell counting kit-8 (CCK-8; Dojindo, Kumamoto, Japan), in accordance with the manufacturer’s instructions. Briefly, 10 μL of WST-8 dye (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt) was added to each well of a 96-well plate [19]. The suspension was incubated for 1 hour and the absorbance was then read at 450 nm with a reference wavelength of 650 nm using a microplate reader (SPECTRAmax 340PC, Molecular Devices, Palo Alto, CA, USA).
Migration assay
The chemotaxis assay was performed in a Boyden chamber system using a transwell with an 8-μm-pore size polycarbonate membrane (Corning, NY, USA). Cells were seeded onto the inner chamber at a density of 8×104 cells per transwell in DMEM with 0.2% FBS and then exposed to aldosterone in the outer chamber for 5 hours [18]. The cells on the inner membrane were then completely removed by wiping with a cotton swab, while the cells on the lower surface of the membrane were fixed in 4% PFA and stained with crystal violet. To count the number of C2C12 cells, the stained C2C12 cells were photographed under cellSens Standard BX53 software (Olympus, Tokyo, Japan), and counted using Image J software (National Institutes of Health, Bethesda, MD, USA).
Measurement of intracellular reactive oxygen species levels
The intracellular reactive oxygen species (ROS) content was measured using the chloromethyl derivative of 2′,7′-dichlorofluorescein diacetate (CM-H2DCFDA, C6827, Invitrogen). Cells were washed with serum-free DMEM and incubated with 10 μM CM-H2DCFDA at 37°C for 30 minutes in the dark at 5% CO2. Then, cells were washed with PBS and visualized using a fluorescent microscope (Carl Zeiss, Jena, Germany). Fluorescence intensity was also read at an excitation wavelength of 490 nm and emission wavelength of 520 nm using a microplate reader (Infinite 200PRO, Tecan Life Sciences, Zürich, Switzerland).
Ligand and receptor binding assay
Recombinant aldosterone was coated onto the wells of Maxisorp 96-well microtiter plates and incubated for 18 hours at 4°C. Each well was washed three times with PBST (0.1% Tween-20 in PBS). Thereafter, the plates were blocked with 1% BSA in PBST for 2 hours. Cell lysates were added to the plates and incubated for 2 hours. The wells were then washed three times [20]. Preparations of the MR antibody (sc-53000, Santa Cruz Biotechnology) in blocking solution were added to the plates and allowed to react at 4°C overnight. After washing, horseradish peroxidase-linked antibody (Cell Signaling Technology) was added and the lysates were incubated for 2 hours; this was followed by five rounds of washes. The reaction was developed with 100 μL of TMB substrate solution and stopped with 100 μL of 1 N H2SO4. The absorbance in the microtiter plates was measured at 450 nm using a microplate reader (Infinite 200 PRO, Tecan Life Sciences).
Statistical analysis
All data are expressed as mean±standard error of the mean from at least three independent experiments with triplicate measurements, unless otherwise specified. The significance of differences among three or more groups was tested by analysis of variance (ANOVA) with post hoc analysis with Tukey’s honest significance test, and differences between two groups were assessed using the Mann-Whitney U test [19]. All statistical analyses were performed using SPSS version 18.0 (SPSS Inc., Chicago, IL, USA). P<0.05 was considered statistically significant.
RESULTS
In vitro effects of recombinant aldosterone on muscle differentiation
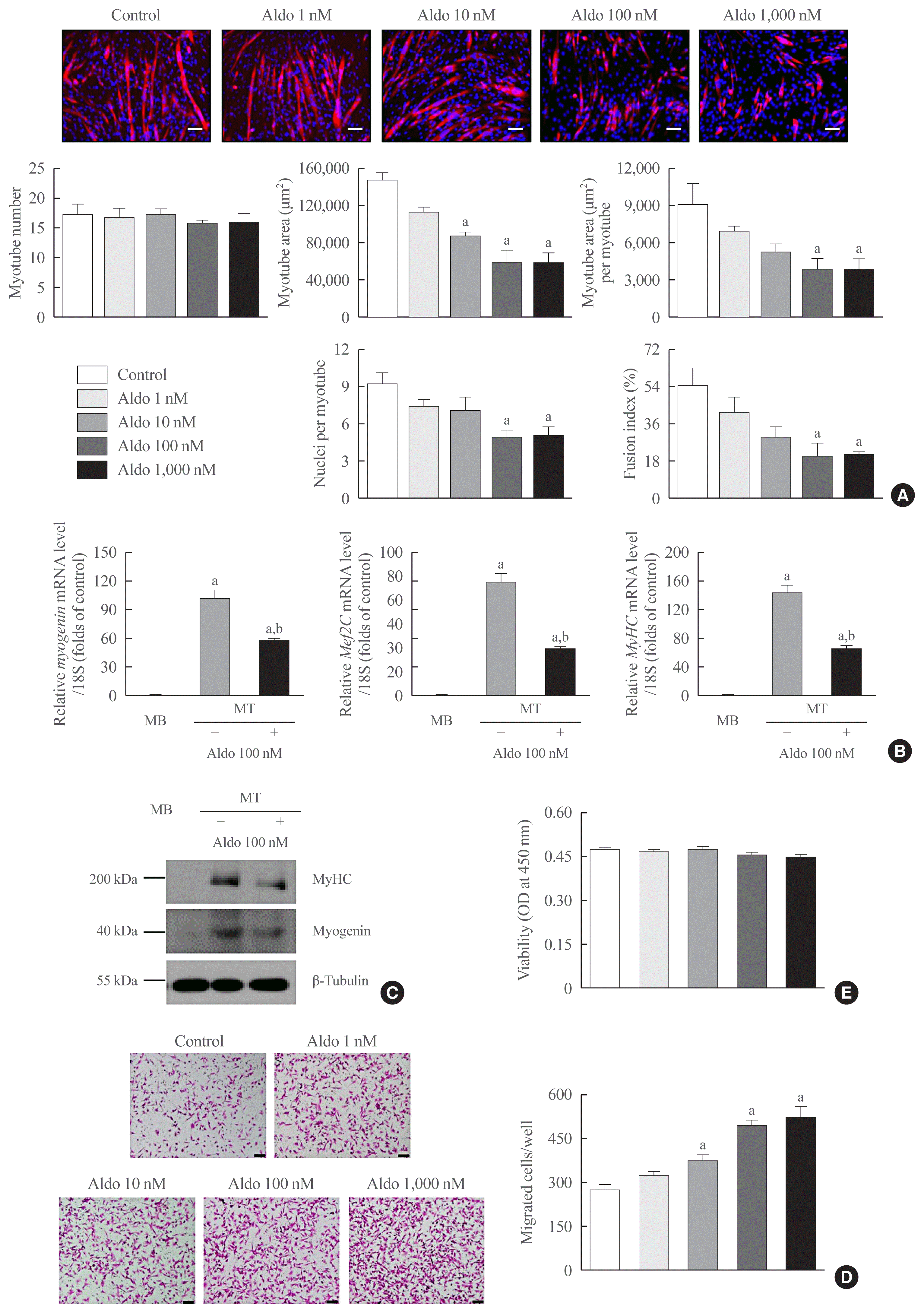
The C2C12 myoblasts were differentiated into mature myotubes in the presence or absence of recombinant aldosterone. Aldosterone markedly suppressed in vitro myogenesis in a dose-dependent manner, as confirmed by the decrease of myotube area, myotube area per myotube, nucleus number per myotube, and fusion index (Fig. 1A). Consistent with these findings, mRNA and protein expression levels of myogenic differentiation markers, such as myogenin, Mef2C, and/or MyHC, were significantly reduced by recombinant aldosterone (Fig. 1B, C, respectively). When we investigated the possible effects of aldosterone on other muscle biology, it was shown to stimulate the migration of C2C12 myoblasts (Fig. 1D) without affecting the viability (Fig. 1E). These results indicate that aldosterone plays a detrimental role in muscle metabolism mainly by inhibiting muscle differentiation.
The inhibitory effects of aldosterone on myogenesis are mediated by increased intracellular ROS
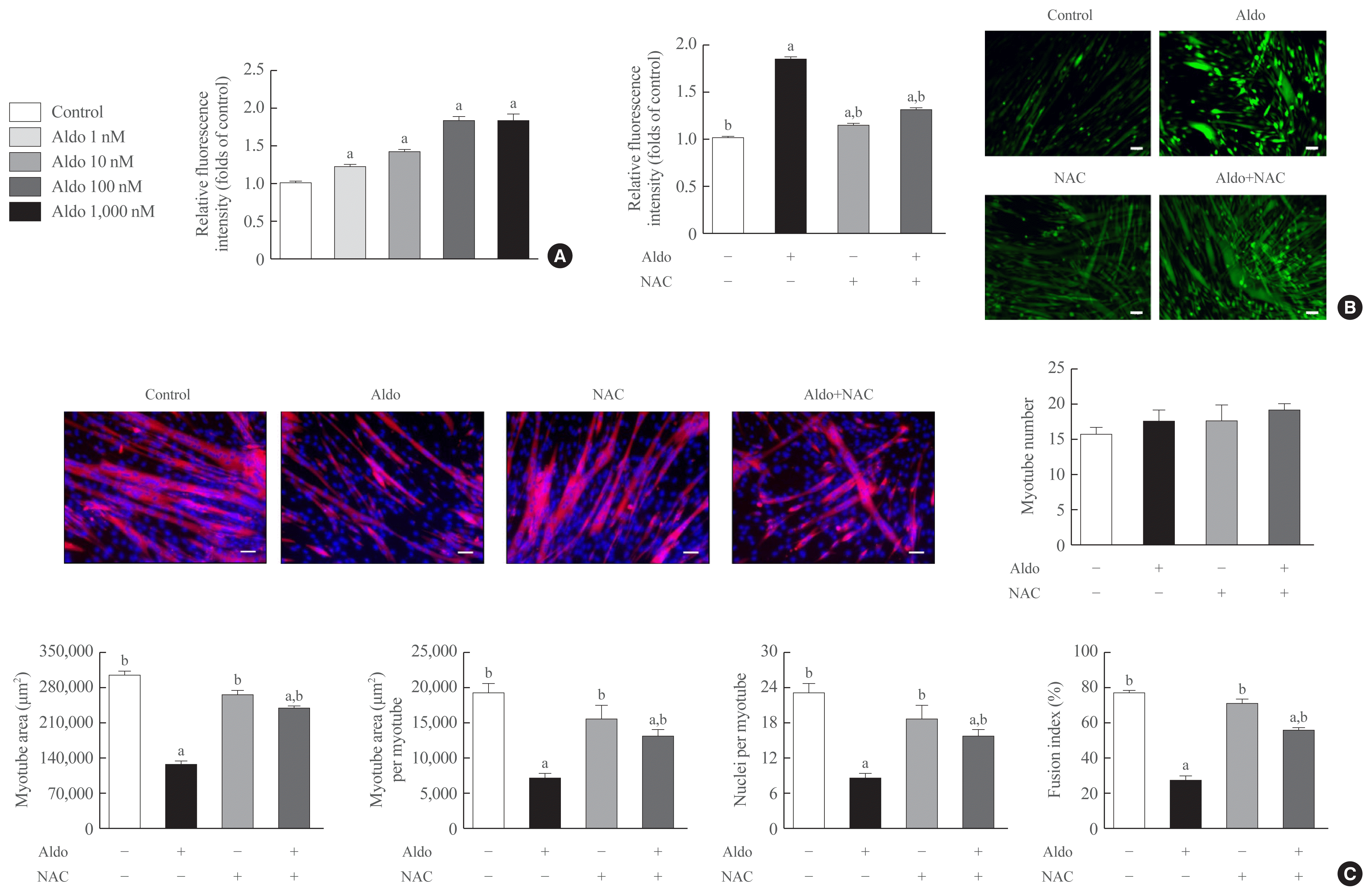
To determine the degree of intracellular ROS generation by recombinant aldosterone, we measured CM-H2DCFDA content in the presence or absence of aldosterone during muscle differentiation. Aldosterone dose-dependently increased intracellular ROS levels in myotubes, and the maximum effect of a 1.9-fold increase was observed at 100 nM aldosterone (Fig. 2A). Interestingly, pretreatment with NAC, a potent biological thiol antioxidant, significantly reduced aldosterone-induced stimulation of intracellular ROS generation in myotubes (Fig. 2B). Furthermore, NAC markedly reversed the decrease of myotube area, myotube area per myotube, nucleus number per myotube, and fusion index due to aldosterone (Fig. 2C), supporting the importance of oxidative stress in the suppression of myogenesis by recombinant aldosterone treatment.
MR is the major receptor for aldosterone in muscle cells
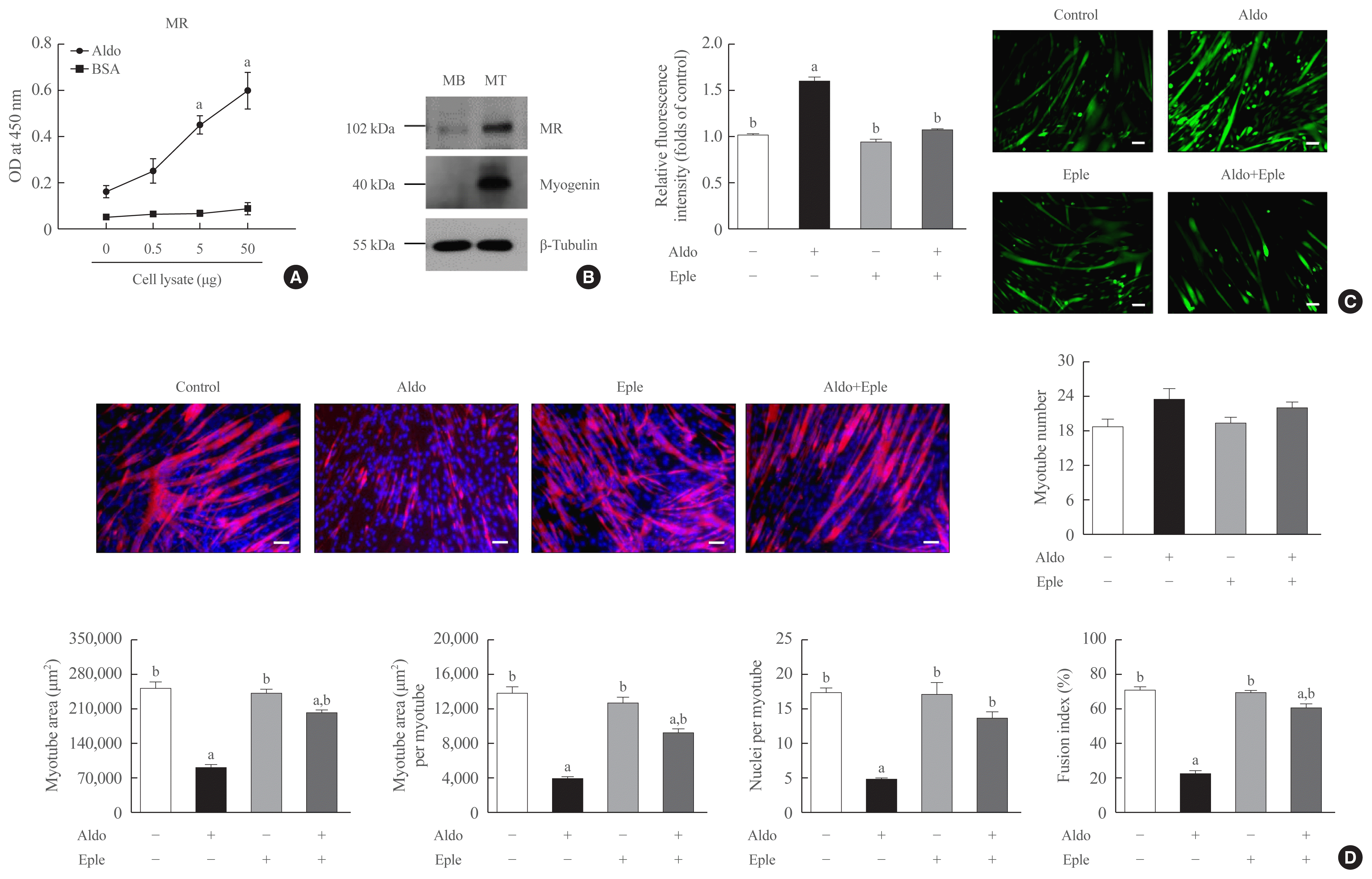
Because MR is known as a receptor for aldosterone in various types of cells [21,22], we focused on the MR mediating the action of aldosterone in muscle cells. A binding affinity experiment using enzyme-linked immunosorbent assay (ELISA) indicated that the amount of aldosterone-associated MR increased as the amount of C2C12 myoblast lysate increased (Fig. 3A). Western blot analyses also showed the increased expression of MR during myogenesis (Fig. 3B). Importantly, pretreatment of C2C12 myoblasts with an MR inhibitor, eplerenone, in differentiation media blocked aldosterone-stimulated intracellular ROS generation (Fig. 3C) and markedly attenuated the suppression of in vitro myogenesis by aldosterone treatment (Fig. 3D). These findings indicate that aldosterone exerts its functions in muscle cells via MR and oxidative stress.
DISCUSSION
We have shown here that aldosterone inhibits in vitro myogenesis by stimulating intracellular ROS generation, which was reversed by NAC treatment. Furthermore, aldosterone and MR were shown to interact in muscle cells, and eplerenone blocked aldosterone-induced oxidative stress and myogenesis. These findings support the hypothesis that hypersecretion of aldosterone, like PA, directly contributes to muscular deterioration and suggest that antioxidants and/or MR antagonists could be an effective therapeutic option to reduce the risk of sarcopenia in these patients.
To investigate the possible mechanisms underlying the detrimental role of aldosterone in muscle metabolism, we focused on changes in redox signaling. In healthy skeletal muscle, the production of oxidant species is matched with the removal by exogenous or endogenous antioxidant molecules [23,24]. However, an imbalance of these processes in certain pathological conditions results in the loss of normal redox equilibrium, which is referred to as “oxidative stress” [25]. Various findings have indicated that the oxidative stress can lead to mitochondrial dysfunction, increase of myonuclear apoptosis and ubiquitin proteasome activity, autophagy deregulation, altered mineral homeostasis, and excitation–contraction uncoupling, all of which are involved in the decrease in muscle mass and quality [23,26,27]. Notably, dysregulation of the renin-angiotensin-aldosterone system (RAAS) damages several organs via oxidative stress mainly from nicotinamide adenine dinucleotide phosphate oxidase-derived ROS overproduction [28–30]. Based on this background, we investigated whether aldosterone increased intracellular ROS production in muscle cells and antioxidants attenuated the impairment of myogenesis due to aldosterone. Our results demonstrated the importance of oxidative stress as a key modulator of the effect of aldosterone on muscle metabolism.
Aldosterone is a final hormone in the RAAS and exerts its effects by binding to MR [20]. Although the interaction between aldosterone and MR is mainly implicated in sodium and potassium homeostasis in the kidneys and the resultant blood pressure control, various tissues have been shown to be under the influence of a local RAAS [31,32]. We also reported that MR was strongly expressed in myotubes, consistent with a previous report [33], and eplerenone completely eliminated the effects of aldosterone on intracellular ROS generation and myogenesis. These results indicate that the interruption of RAAS via MR antagonists would be beneficial to maintain muscle health, in addition to the known role in reducing cardiovascular risks [10,11].
Several points require discussion for the appropriate interpretation of our data. Firstly, although intracellular ROS levels in myotubes were dose-dependently increased by aldosterone treatment from 1 nM (36 ng/dL), which is the typical aldosterone concentration difference in subjects with and without PA, maximal detrimental effects on muscle biology were observed at 100 nM (3,600 ng/dL) aldosterone; therefore, using 100 nM aldosterone was appropriate for our study. Similarly, we acknowledge inevitable differences between in vitro environments and human systems. Secondly, spironolactone binds not only to MR but also to progestogen and androgen receptors [34]; therefore, to adequately investigate links between aldosterone and MR in muscle, we used eplerenone, a more selective MR antagonist than spironolactone. Thirdly, our results showing increased migration of C2C12 myoblasts, despite suppressed muscle differentiation, by aldosterone were interesting. While we speculate that this could be an effect to compensate for damaged muscle cells by aldosterone, specific mechanisms and implications must be elucidated in future work. Lastly, angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are widely used in clinical practice to abolish the effects of RAAS [10]. Considering these medications may partially reduce blood aldosterone levels, the protective role of ACE inhibitors and ARBs in human muscle health is a highly interesting research topic.
As the clinical importance of sarcopenia grows with the progressing aging of society, many studies are being conducted to find and reverse the related risk factors. PA is attracting attention not only as the most common cause of secondary hypertension, but also for the adverse outcomes in musculoskeletal systems [13,35,36]. In the present study, we demonstrated that aldosterone leads to reduced myogenesis by directly binding to its receptor and the resultant increase of intracellular ROS production in muscle cells. These results provide the possible underlying mechanism explaining the deleterious effects of excess aldosterone on muscle metabolism. More interventional clinical studies demonstrating the improvement of muscle health by adrenalectomy, antioxidants, or MR antagonist in PA are required to translate the experimental results to humans.
 XML Download
XML Download