

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Prolonged autonomous cortisol secretion leads to Cushing syndrome (CS). Endogenous CS is a rare disease with two to three cases per million per year [1]. In full-blown CS signs and symptoms accumulate to the characteristic Cushing phenotype including skin changes, such as rubeosis, acne, ecchymosis, purple striae, and skin infections. Patients also have thin arms and legs and increased intraabdominal, truncal, neck and facial fat accumulation and various other symptoms [2,3]. Co-morbidities include cardiovascular, metabolic, psychiatric, and musculoskeletal disease. The latter includes osteopenia, osteoporotic fracture, sarcopenia, and myopathy. Thus, CS is a special form of sarcopenic obesity [4]. Muscle weakness is one of the frequent symptoms reported by affected patients [3]. Its outcome following remission of CS remains dissatisfying, and effective treatment options are currently lacking. This review will summarize the current knowledge about epidemiology, pathophysiology and long-term outcomes of Cushing-associated myopathy.
Go to : 
SEARCH STRATEGY AND SELECTION CRITERIA
I searched MEDLINE (1932 to 2020) using the search terms “Cushing syndrome,” “Cushing’s syndrome,” “Cushing’s disease,” “Hypercortisolism,” and “myopathy.” In addition, I searched with the terms ectopic adrenocorticotropic hormone (ACTH) syndrome, ectopic Cushing’s syndrome and ectopic Cushing syndrome. I largely selected publications in the past 5 years but did not exclude commonly referenced and highly regarded older publications. I also searched the reference lists of articles identified by this search strategy and selected those I judged relevant. Review articles are cited to provide readers with more details and more references.
Go to :
CUSHING PHENOTYPE, SUBTYPES, CO-MORBIDITIES
Eighty percent of the cases are caused by an ACTH-dependent CS, mostly by corticotroph pituitary adenomas (Cushing disease), or rarely by ectopic ACTH producing neuroendocrine tumors. The remaining 20% are caused by ACTH-independent adrenal CS [5]. This includes cortisol producing adenomas, rarely carcinomas of the adrenal cortex, and bilateral hyperplasia forms. Late diagnosis of CS is associated with high morbidity and mortality [6]. Due to a strong predominance of women in pituitary and adrenal CS, the ratio of female to male patients is ≥3:1, with mortality rates higher in females than in males [7], and untreated with a mean life expectancy of 5 years [8]. Clinically, CS is characterized by the consequences of a long-lasting increase in plasma cortisol levels, which manifests itself as a multi-system disease in virtually all organ systems due to the universal expression of the glucocorticoid receptor. The dominant effects are on glucose and fat metabolism, the immune and the musculoskeletal system. Furthermore, the central nervous system and the cardiovascular system are affected [9]. Chronic excessive cortisol exposure in patients with CS determines severe physical morbidity and psychological dysfunctions, which invariably impair health related quality of life [9–13]. By comparison, CS patients have worse quality of life compared to patients with other pituitary diseases [14–16]. Although quality of life improves after successful treatment of hypercortisolism, it does not completely normalize [15,17–24]. This is likely due to the persistence, to various degrees, of several features associated with previous cortisol excess, including cardiovascular morbidity, myopathy-related fatigability, bone fragility, affective alterations and cognitive dysfunctions [16,25,26]. All these factors, along with negative disease perception, affect well-being of CS patients, even years after remission [11,12]. Whereas endogenous CS is rare, up to 3% of the general population is taking prescribed glucocorticoids [27], and their side effects represent a considerable clinical burden [28].
Go to :
CUSHING’S SYNDROME-ASSOCIATED MYOPATHY: EPIDEMIOLOGY
Preservation of muscle function is a main determinant in health and disease [29], whereas impaired muscle function and sarcopenia are associated with frailty, impaired health related quality of life and increased mortality [30]. Hypercortisolism associated myopathy is one of the most prominent clinical features of CS which has been studied so far only in small series of patients using electromyography, histology, imaging and functional testing [31]. Muscle weakness is reported by patients with florid CS in 40 to 70% and it appears to be more pronounced in females [3]. Systematic clinical testing, however, identifies myopathy in nearly every patient. The most commonly impaired compartment is the proximal musculature of the lower limbs. Accordingly, patients typically complain about the inability to get up from a squatting position or to climb stairs whereas running or walking is less frequently affected. Steroid induced myopathy is characterized by a decrease in muscle fiber conduction velocity on electromyography [32] and histology reveals an atrophy of type IIa muscle fibers (Fig. 1) [33]. The natural course of CS associated myopathy gave conflicting results, ranging from limited improvement to persistence despite remission of hypercortisolism [31,34]. However, clinically restitutio at integrum has not been demonstrated. The long-term impairment of muscle dysfunction and reduced responsiveness to physical activity of normal life suggest that the understanding of its pathophysiology is essential to develop appropriate prevention and intervention strategies. In a clinical study in 10 patients with CS, Minetto et al. [35] showed lower serum creatine kinase and plasma myoglobin levels compared to controls. Electrophysiologic studies demonstrated decreased muscle fiber conduction velocity and myoelectric manifestations of fatigue in all muscles [35]. In a short-term study by the same authors five healthy males received 8 mg dexamethasone orally for 5 days followed by muscle biopsies [36]. The study demonstrated that both type 1 and type 2A fibers decreased significantly by 11% and 17%. In a case report of adrenal CS causing steroid myopathy the authors used quantitative muscle ultrasonography for the assessment of glucocorticoid-induced changes in muscle mass and structure. The patient had low muscle strength, low physical performance, and low muscle mass both preoperatively and postoperatively [37]. Table 1 is summarizing the clinical findings.
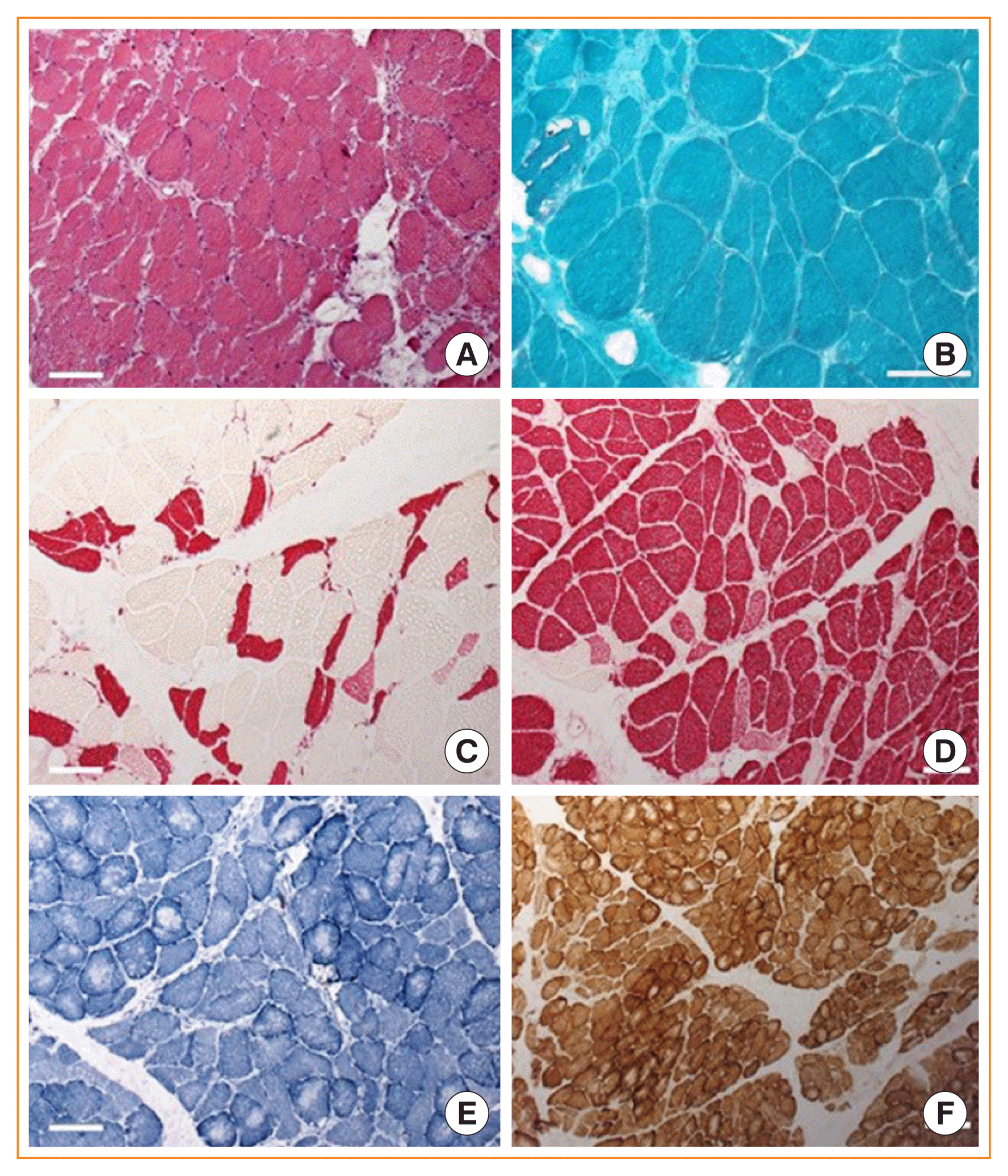 | Fig. 1Biopsy of left musculus gastrocnemius of a patient with florid Cushing syndrome. (A) H&E stain reveals disseminated atrophic muscle fibres with mild fibrosis and increase in connective tissue. Myonucleii are somewhat more internalised and single fibres show subsarcolemmal vacuolization. (B) Trichrome Gömori staining displays no protein aggregation or ragged red fibres. Anti-myosin fast (C) and slow (D) stainings show a pronounced type-2 fibre atrophy; however, some type-1 fibres are also atrophic. Oxidative stainings (E, nicotin amide adenine dinucleotide [NADH]; F, cytochrome C oxidase/succinate dehydrogenase [COX/SDH] double staining) shows central reduction of oxidative enzymes in some fibres, pointing at an energy level alteration in those myofibres (×25).
|
Table 1
Signs, Symptoms and Biochemical and Other Abnormalities Associated with Myopathy in Cushing Syndrome
| Symptom | Reference |
|---|---|
| Self-reported muscle weakness (in 40%–70 %) | [3] |
| Reduced grip strength | [31] |
| Prolonged chair rising test time | [31] |
| Impaired quality of life associated with low muscle strength | [34] |
| Low serum creatine kinase | [35] |
| Low plasma myoglobin levels | [35] |
| Lower IGF1 levels associated with worse myopathy | [63] |
| Decreased muscle fiber conduction velocity | [36] |
| Decrease of type 1 and type 2A fibers (11% and 17%) | [36] |
![]()
Patients with CS also have increased cardiovascular morbidity and mortality despite successful therapy [7,38–40]. This increased cardiovascular risk is also evident in patients on long-term glucocorticoid therapy [41]. Opposite to the atrophy of the peripheral musculature, patients with endogenous CS show an increased prevalence of left ventricular hypertrophy with concentric remodeling and left ventricular systolic and diastolic dysfunction [42–44]. A restricted right ventricular function could also be shown magnet resonance morphologically [45]. Myocardial fibrosis has also been observed in patients with florid CS [46]. Whether these structural changes are reversible after therapy is a matter of controversy, the pathophysiology is largely unclear. Persistence of the cardiac dysfunction despite successful treatment is suspected [47]. Cardiac remodeling appears to be only partially explainable by the presence of arterial hypertension in patients with CS. Previous studies showed no correlation between the degree of left ventricular hypertrophy and blood pressure; the development of hypercortisolism-associated cardiomyopathy with typical structural changes was also observed in Cushing patients without arterial hypertension [42,43,48]. Left ventricular hypertrophy seems to be primarily dependent on the duration of the hypercortisolism [43]. The direct effects of prolonged hypercortisolism on the myocardium are poorly understood [40,49]. Myocardial biopsies from patients with CS indicate myocardial hypertrophy, fibrosis, and myofibrillolysis [50]. The increased expression of atrogin-1, a marker of muscle atrophy, was observed [51,52].
Go to :
PATHOPHYSIOLOGY OF HYPERCORTISOLISM-ASSOCIATED PERIPHERAL MYOPATHY
Peripheral muscle mass is tightly controlled through the regulation of protein metabolism, myoblast proliferation, and myocyte differentiation – all vital processes in the repair and maintenance of healthy muscle tissue. In muscle, mammalian target of rapamycin (mTOR) is a central regulator of protein synthesis, regulating numerous components including the initiation and elongation factors [53]. The converse, protein degradation is regulated by components of the E3 ubiquitin proteosomal system (UPS), including muscle-ring finger protein-1 (MuRF1) and atrogin-1, both target cellular proteins to the proteasome for hydrolysis [54,55]. It is well established that glucocorticoids drive muscle atrophy through modulation of protein metabolism [56–58]. The precise molecular mechanisms underpinning glucocorticoid action in the regulation of intramyocellular protein metabolism and myoblast proliferation have been recently explored in a couple of rodent studies. In ACTH-infused rats, as a chronic model of hypercortisolism, it was shown that the forkhead box O3a (FOXO3a) promoter is a target of the activated glucocorticoid receptor, leading to upregulation of FOXO3a, MuRF1, and atrogin-1 mRNA. The glucocorticoid receptor antagonist RU486 significantly decreased transcriptional activity and expression of FOXO3a. Treatment with RU486 also reduced MuRF1 and atrogin-1 expression in accordance with reduced enrichment of FOXO3a and polymerase II on the respective promoters. Knockdown of FOXO3a prevented dexamethasone-induced MuRF1 and atrogin-1 expression. These results indicate that FOXO3a plays a pivotal role in hypercortisolism induced muscle atrophy through modulation of MuRF1 and atrogin-1 [59].
In a study using murine C2C12 myotube cultures, the main rodent glucocorticoid corticosterone decreased myotube area, decreased protein synthesis, and increased protein degradation [60]. This was paralleled by decreased mRNA expression of insulin-like growth factor 1 (IGF1), decreased activating phosphorylation of mTOR, decreased phosphorylation of 4E binding protein 1, and again increased mRNA expression of key atrophy markers including atrogin-1, FOXO3a, myostatin, and MuRF1. The authors further explored the role of local 11β-hydroxysteroid dehydrogenase-1 activity which converts inactive glucocorticoids into corticosterone. Selective 11β-hydroxysteroid-dehydrogenase-1 inhibition blocked the decrease in protein synthesis, increase in protein degradation, and reduction in myotube area induced by dehydrocorticosterone, the inactive glucocorticoid converted into active corticosterone. These data underscore the role of local glucocorticoid activation by 11β-hydroxysteroid-dehydrogenase and potential use of selective 11β-hydroxysteroid-dehydrogenase 1 inhibitors to ameliorate muscle-wasting effects associated with glucocorticoid excess.
Finally, the adaptations of different muscle groups of C57BL/6 female mice were studied after daily intraperitoneal injections of high dose dexamethasone in a chronic glucocorticoid excess model. Gene expression of major components of the intracellular signalling pathways controlling muscle mass and metabolism demonstrated a significant muscle fibres atrophy after 15 days, associated with enhanced gene expression of Murf1 (3 to 6-fold changes) and myostatin (6 to 20-fold change) [61].
Go to :
CUSHING MYOPATHY IN THE GERMAN CUSHING’S REGISTRY
Since its foundation in 2012, the German Cushing Registry has succeeded in registering more than 900 deeply phenotyped patients with CS in 10 centers. Three hundred and fifty-five subjects who were evaluated for possible CS but were found unaffected (so called rule-out Cushing cases) are also registered and phenotyped as controls. The registry is closely linked to a comprehensive biobank (with plasma, DNA, tumor tissue, urine, saliva, stool, hair). The German Cushing Registry is hypothesis driven and aims to characterize the musculoskeletal comorbidities of CS patients. This has resulted in four publications since 2015 covering clinical, biochemical, and genetic aspects related to this topic [31,34,62,63].
Since Cushing-associated myopathy represents a relevant comorbidity, we evaluated its severity in a cross-sectional study of 198 patients with CS and 93 controls. This was the first study of its kind using standardized upper limb muscle function measurements assessed by a hydraulic hand dynamometer (Jamar, JLW Instruments, Chicago, IL, USA) for grip strength and lower limb function using the chair rising test. Active CS patients showed significantly lower grip strength than control subjects, demonstrating 84% versus 98% of age and gender corrected grip strength on the non-dominant hand. Similarly, the average chair rising test time was higher in patients with CS. By applying multiple regression analysis, diagnosis of CS and age contributed to the performance of the chair rising test, but not gender or body mass index [31].
In a consecutive longitudinal study, we investigated the long-term outcome of muscle function in 88 patients at time of diagnosis and thereafter following surgical remission in annual intervals, over a period of up to 4 years [34]. As in our previous study, grip strength was decreased to 83% of normal controls at time of diagnosis. Following surgical remission, grip strength decreased paradoxically to 71% at 6 months and showed no improvement during further follow-up. In multivariate analysis, we identified age, waist-to-hip ratio, and glycated hemoglobin1A as independent risk factors. Furthermore, impaired muscle strength was strongly correlated with low quality of life.
In a multi-centric study, we evaluated the impact of three well-characterized glucocorticoid receptor polymorphisms (BclI, N363S, ER22/23EK, and A3669G), which are known to influence peripheral glucocorticoid sensitivity. Peripheral blood leukocytes glucocorticoid receptor genotyping and muscle function was analysed in 205 patients with proven endogenous CS and 125 control [62]. In florid CS, normalized handgrip strength of the dominant hand was higher in A3669G minor allele than in wildtype carriers, an effect which was not seen in patients with CS in remission or ruled-out CS patients. These data may explain inter-individual differences of glucocorticoid-induced myopathy in patients with endogenous CS.
Finally, we analysed the impact of individual serum IGF1 concentrations on long-term myopathy outcome in CS. In a prospective longitudinal study of 31 patients with florid CS, we analyzed IGF1 and IGF binding protein 3 concentrations at the time of diagnosis and following surgical remission over a period of up to 3 years [63]. The results show that individual serum IGF1 concentrations in the postoperative phase were strongly predictive of long-term grip strength outcome, and lower IGF1 concentrations were associated with a lower muscle mass at three years. In contrast, patients with high IGF1 standard deviation (SD) scores showed an improvement in grip strength within the follow-up period (P=0.009), whereas patients with low IGF1 SD scores had an adverse outcome with persisting muscle dysfunction. These data argue strongly for an essential role of IGF1 for muscle regeneration in the phase after correction of hypercortisolism.
Go to :
EXOGENOUS CORTICOSTEROID PHARMACOTHERAPY AND MYOPATHY
Synthetic glucocorticoids, such as dexamethasone and prednisolone, belong to the top 460 list of essential medicines of the World Health Organization (https://www.who.int/groups/expert-committee-on-selection-and-use-of-essential-medicines/essential-medicines-lists). Due to their multitude of actions, they are used in very different indications. These range, among others, from immunosuppressive effects in the oncologic or allergic setting to anti-inflammatory actions in autoimmune joint, liver, gastrointestinal or connective tissue diseases, asthma, chronic obstructive pulmonary disease (COPD), or most recently severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) associated pneumonia. Epidemiologic studies estimate that the annual prevalence of glucocorticoid use is 3% in Western societies [27]. This number has not changed in the last 2 decades. Glucocorticoid use is age-depending: it increases to 7% in persons between 60 and 79 years, and to >10% in those 80 years or older [27]. In asthma patients its use is dose-dependently associated with increasing morbidity and mortality [64].
Patients with COPD often report limb muscle dysfunction during exacerbation. Contributing factors leading to muscle dysfunction are similar to those seen in stable COPD and include glucocorticoid use [65]. However, the correlation between glucocorticoid dose and quadriceps strength is not straightforward [66]; thus, raising the possibility that the effects may be an interaction between the steroids, immobility, and/or inflammation and not simply a steroid effect alone. In stable patients with COPD, a 2-week course of 30 mg prednisolone daily did not cause significant skeletal muscle dysfunction or alter metabolic parameters during exercise [67]. A recent systematic review analysed the effect of glucocorticoid pharmacotherapy on inspiratory muscle function and proximal limb myopathy of asthma patients. Of 11 included studies two reported significant associations between dosage of oral glucocorticoid use and inspiratory and limb muscle function. Eleven to 36% of patients receiving glucocorticoids intravenously because of acute exacerbation suffered from limb muscle weakness during or after critical care admissions. In summary, because of the different doses used and the heterogeneity of the underlying clinical condition, the pattern of muscle damage and dysfunction appears to be more heterogenous in exogenous forms of CS than in endogenous forms.
Go to :
CONCLUSIONS
In summary, the presented studies demonstrate that proximal myopathy is a frequent and clinically relevant co-morbidity of CS. Age, waist-to-hip-ratio and a hyperglycemic metabolic state are the main risk factors. Following biochemical remission in response to successful pituitary or adrenal tumor resection, there is consistently in several of our studies an early worsening of myopathy, and during long-term follow-up a partial recovery. Impairment of quality of life is closely linked with persistence of myopathy. There appears to be a genetic impact of glucocorticoid polymorphism on the degree of individual myopathy. This might explain to some extent, why myopathy has such a broad intra-individual variability. Finally, our data demonstrate that an intact postoperative growth hormone (GH) IGF system is essential for recovery of myopathy. In the presence of a lack of direct evidence from therapeutic studies, one might speculate that GH replacement therapy may be beneficial in those individuals who have a persistently low IGF level, and who do not respond to classical GH stimulation tests. Whether targeted physiotherapy in the postoperative period is able to improve myopathy, is currently investigated in our institution in a prospective randomized trial.
Go to :
 XML Download
XML Download