

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Regulation of food intake and energy homeostasis is tremendously complex, and recent findings have revealed that the motivation for food is closely linked to our brain reward system, which responds to the hedonic properties of food and may interact with homeostatic regulation for integrative control of feeding behaviors. While the homeostatic regulation of food intake can coordinate the energy balance between energy intake and expenditure, palatable food can stimulate food intake even when hunger is absent, and this motivation to obtain palatable food can override homeostatic signals [1–3]. It has been suggested that the brain’s reward systems mediate the motivation toward palatable food, which involves learning associated with sensory perception, such as the sight, smell, and taste of food, as well as cues that are associated with food or the hedonic context of food intake. The dopamine reward system is the most prominent reward circuit in the brain, and can control appetite and motivational feeding behavior [1–3].
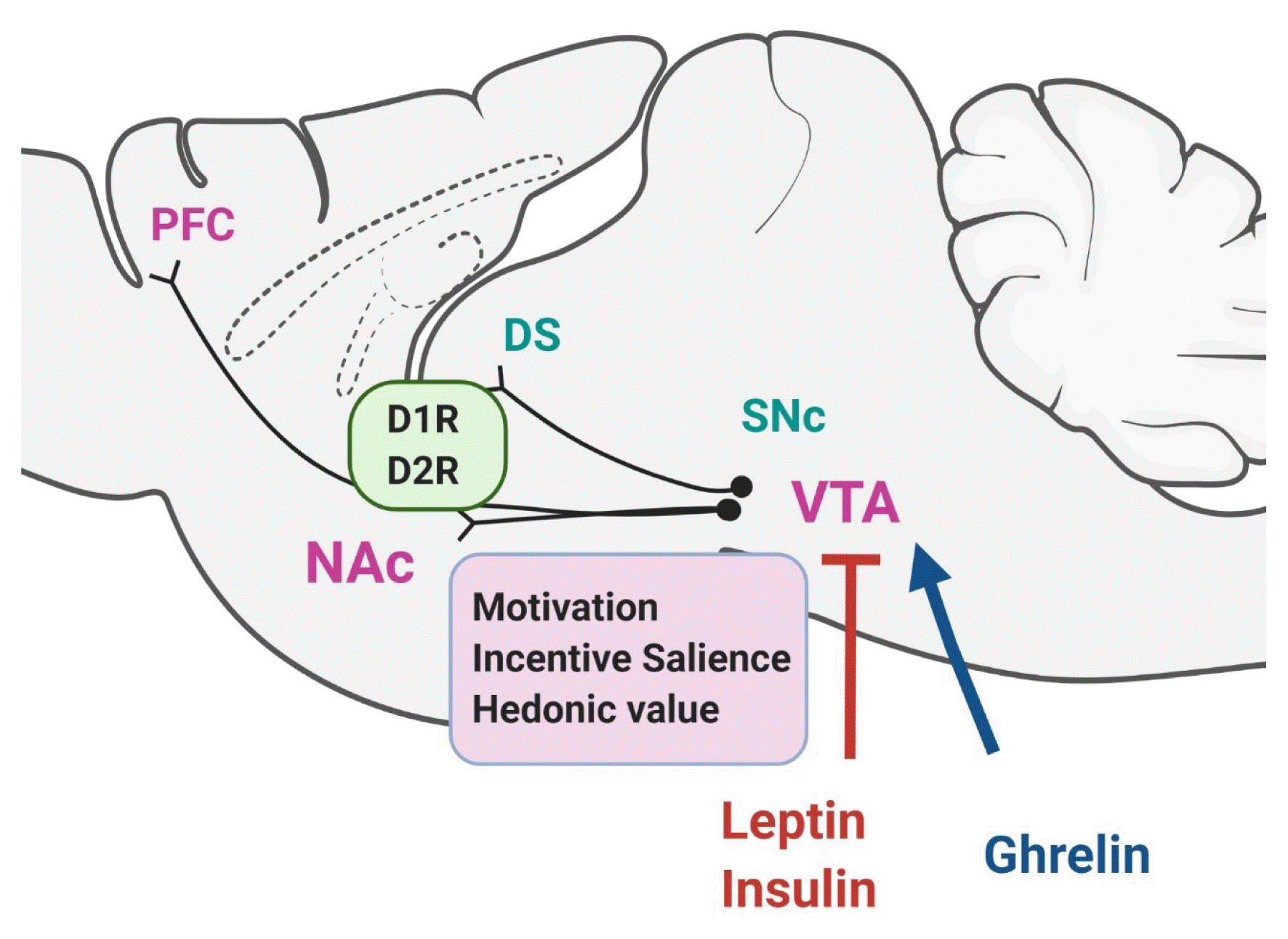
Dopamine is synthesized mostly by midbrain neurons in the substantia nigra and the ventral tegmental area (VTA). Dopaminergic neurons can be identified by immunohistochemistry for tyrosine hydroxylase, the rate-limiting enzyme of dopamine synthesis, and are designated as group A cells (cells containing catecholamines, primarily noradrenaline and dopamine); specifically, the dopamine-containing cell groups are A8 through A16 [4–6]. The nigrostriatal pathway constitutes the projection of neurons from the A8 cells, which are located in the retrorubral field, and the A9 neurons in the substantia nigra pars compacta to the dorsal striatum (DS). The nigrostriatal pathway is primarily involved in the control of movement execution and goal-directed behaviors, including reward-related cognition. The mesolimbic and mesocortical pathways are other major dopaminergic pathways originating from the VTA. The A10 cell group from the VTA projects to the ventral striatum, which is known as the nucleus accumbens (NAc), and also to the prefrontal cortex and other limbic areas. This pathway plays a crucial role in reward-related behaviors and motivation. The tubero-infundibular pathway comprises the arcuate nucleus (cell group A12) and periventricular nucleus (cell group A14) of the hypothalamus projecting to the pituitary, and is known to control the secretion of pituitary hormone and prolactin [4–6]. The effect of dopamine is mediated by the binding of dopamine to the dopamine receptors, which belong to a family of seven transmembrane domain G-protein-coupled receptors. Five different subtypes of dopamine receptor have been identified based on their structural and G-protein coupling properties: the D1-like receptors (D1 and D5), which stimulate intracellular cyclic adenosine monophosphate (cAMP) levels, and the D2-like receptors (D2, D3, and D4), which inhibit intracellular cAMP levels [3].
The mesolimbic and mesocortical pathways in the dopamine reward system have attracted considerable attention regarding the control of food intake in association with hedonic feeding. Mounting evidence suggests that reward-related motivational food intake interacts with the homeostatic system of energy balance, and it appears that the dopamine system plays a critical role in the control of feeding behavior, both through the reward-related circuit and by contributing to the homeostatic regulation of energy balance by participating in the hypothalamic circuit of food intake [7]. Herein, I will review and discuss current findings on the role of the dopamine system in the control of food intake through the reward-related circuit and the homeostatic hypothalamic system.
DOPAMINERGIC CONTROL OF FOOD INTAKE THROUGH THE REWARD CIRCUIT
Initial studies demonstrated that as like addictive drugs, palatable foods that are rich in fat and sugar content can significantly activate the dopamine reward system. For example, when dopamine was measured by microdialysis in the NAc of freely moving rats, it was observed that extracellular dopamine in the NAc increased significantly during and after receiving food as a reward [8]. Roitman et al. [9] measured dopamine throughout the entire sequence of feeding behavior by sampling dopamine every 100 ms, using fast-scan cyclic voltammetry in the NAc of rats trained to press a lever to obtain sucrose, and found that subsecond dopamine signaling in the NAc operated as a real-time modulator of food-seeking behavior. In contrast, intra-oral delivery of quinine, which has an aversive taste, suppressed dopamine release in the NAc, showing that dopamine signals were differentially modulated by appetitive (rewarding) and aversive stimuli [10].
Based on the role of the dopamine reward system in food-seeking behavior, considerable evidence has revealed that there is an interplay between the homeostatic regulator and dopamine system, such that homeostatic regulators of food intake interact with the dopamine reward system to exert an inhibitory or enhancing effect on food intake. For example, it has been demonstrated that leptin and insulin inhibit VTA dopamine neurons, while ghrelin activates them [11–17]. Hommel et al. [11] established that VTA dopamine neurons express the leptin receptor, and in response to leptin, these VTA leptin receptors are activated and suppress the activity of dopamine neurons. Administration of leptin to the VTA was found to decrease food consumption, while knockdown of leptin receptors in the VTA resulted in an increase in food intake, locomotor activity, and hedonic feeding [11]. Fulton et al. [12] observed a similar result, showing that leptin-responsive VTA dopamine neurons project to the NAc; moreover, in leptin-deficient ob/ob mice, leptin function in the VTA-NAc circuit was altered, indicating that leptin can modulate the dopamine mesolimbic reward pathway. In parallel, Farooqi et al. [13] reported that using functional magnetic resonance imaging, in patients with congenital leptin deficiency, leptin treatment diminished perceptions of food reward, which was associated with the modulation of neural activation in the NAc-caudate region, suggesting that leptin reduced the food reward by acting on the dopamine reward system [13,18]. However, it appears that leptin receptor expression is very low in the VTA; therefore, whether in vivo leptin can physiologically and significantly inhibit the VTA dopamine neuron activity through its receptors remains uncertain (Fig. 1) [1,3].
Consistent with the above concern, conditional knockout (KO) mice lacking functional leptin receptor selectively in dopamine neurons, using dopamine transporter (DAT) promoter-driven Cre-transgenic mouse line, exhibited normal body weight and hedonic feeding responses [19]. Instead, leptin receptor signaling in midbrain dopamine neurons appears to be important for the modulation of anxiety-related behaviors [19]. Evans and Anderson [20] recently reported an assessment of the functional importance of leptin receptors in midbrain dopamine neurons by exclusively expressing leptin receptors in dopamine neurons in otherwise leptin receptor-deficient mice to test whether dopamine neuron-restricted leptin signaling is effective for improving the obesity phenotype of leptin receptor-deficient mice. In these mice, where leptin receptor was expressed exclusively in dopamine neurons, the obesity phenotype was not ameliorated; rather, male mice developed an exacerbated obesity phenotype, although these mice still showed an acute leptin-induced effect on food intake [20].
Leinninger et al. [21] reported another circuit where leptin acts via leptin receptor-expressing GABAergic lateral hypothalamic neurons, and then modulates the mesolimbic dopamine system and suppresses feeding. Recent evidence suggests that optogenetic activation of lateral hypothalamus GABA neurons that project to the VTA can modulate feeding, food seeking, and reward [22,23]. These findings therefore underscore the importance of the VTA-to-NAc reward circuit as a mediator of food-seeking behavior, as well as the indirect action of leptin on the dopamine reward system for control of feeding.
However, when dopamine-deficient mice were generated, these mice were aphagic and stopped feeding a few weeks after birth, ultimately dying of starvation by 4 weeks of age; restoration of dopamine production of the caudate-putamen (DS), but not of the NAc, restored feeding [24]. Other studies have also revealed the importance of the DS, which is involved in goal-directed behavior including reward-related cognition and learning, rather than the NAc, within the dopamine circuit in the control of motivational feeding behavior. For example, injection of a dopamine antagonist into the DS of rats, but not into other brain areas such as the NAc, amygdala, or frontal cortex, produced a reduction in food reward-associated lever pressing [25]. A study reported by Johnson and Kenny [26] revealed that animals given a “cafeteria diet” (i.e., a palatable high-fat diet) gained weight through compulsive eating behavior and had decreased dopamine D2 receptor expression in the striatum. In another recent study, Tellez et al. [27] found that dorsal and ventral striatum (NAc) in mice are differentially involved in the hedonic and nutritional actions of sugar. Dopamine release was increased in the DS only when energy was present in a sweet solution, indicating that it encoded the nutritional value, whereas the NAc was responsible for the hedonic value of sugar solution [27]. Thus, it appears that there are separate circuitries in the striatum that are differentially recruited to the hedonic and nutritional values of sugar, allowing the animal to sense nutrients for feeding [27]. On the other hand, it has been reported that dopamine in the DS circuit can enhance the incentive salience of food reward cues, especially when foods are both caloric and palatable, thus implying that DS plays a role in “incentive motivation” (Fig. 1) [28,29].
Brain imaging of the dopamine reward system and human obesity
Numerous studies of humans have investigated the relationship between dopamine reward system activation and eating behaviors, mostly using positron emission tomography (PET) and single photon emission computed tomography to measure baseline binding potential (BP) for dopamine receptor D2R (D2/D3R) ligands.
Small et al. [30] reported a correlation between the reduction in binding of raclopride, a dopamine ligand, and meal pleasantness in the DS, but not in the NAc, in a PET study of healthy human subjects. Consistent with this finding, numerous studies reported that striatal dopamine D2R availability was reduced in obese individuals in proportion to their body mass index (BMI), suggesting a negative correlation between D2R BP and BMI [31–37]. This reduced D2R BP measured by PET analysis is interpreted as indicating increased dopamine release due to competition between endogenous dopamine and the radioligand for binding to D2R.
However, other reports have described positive correlations between D2R BP and a higher BMI [38,39]. Dunn et al. [38] measured D2R availability using PET and fallypride, a high-affinity dopamine D2/D3R antagonist radioligand, in lean (n=8) and obese (n=14) females. In the caudate, there was a positive association of BMI and leptin with D2R availability and D2R availability was increased in obese subjects, indicating reduced dopamine release competing with the radioligand [38]. Caravaggio et al. [39] found that in individuals with a normal range of BMI, there was a positive correlation between BMI and D2R agonist binding in the NAc, but no correlation with D2R antagonist binding; furthermore, no correlation with either D2R agonist or antagonist binding was found in the DS.
However, other studies reported no significant difference in D2R availability according to BMI. For example, Eisenstein et al. [40] found no correlation between striatal D2R BP and BMI values using the D2R antagonist. Karlsson et al. [41] also reported that there were no significant differences in D2R availability revealed by raclopride between obese and nonobese subjects in any brain region.
Interestingly, Guo et al. [42] reported that opportunistic eating and obesity are positively associated with D2R BP using fallypride in the DS, which appears to be associated with habit formation, while there was a negative correlation between BMI and D2R BP in the NAc. The finding of distinct patterns in the DS and the NAc in a D2R brain imaging study provides further support for the possibility that obese people might have alterations in the dopamine system, with increased susceptibility to opportunistic overeating but a decrease in reward in response to food intake; this alteration in neural circuitry could be an important contributor to behavior in the pathogenesis of obesity [42].
To better understand whether changes in dopamine signaling are a cause or a consequence of an increase in body weight, several groups have studied D2R binding by PET in obese individuals who underwent bariatric surgery, although these studies were limited by a small sample size. Dunn et al. [43] reported that dopamine D2R availability decreased at 7 weeks after surgery (roux-en-Y gastric bypass [RYGB] or vertical sleeve gastrectomy surgery, five females) in the DS, as well as in the NAc and the hypothalamus, and these decreases were accompanied by significant decreases in plasma insulin and leptin levels. In contrast, Steele et al. [44] observed that D2R availability increased 6 weeks after gastric bypass surgery (laparoscopic RYGB, five females), and this increase appeared to be proportional to the amount of weight lost, thus showing opposite results, probably due to the small number of individuals in the study. With a relatively larger sample (n=18), de Weijer et al. [45] reported that 6 weeks after RYGB surgery, BMI significantly reduced, but there was no correlation between BMI and D2R (D2/3R) BP before or after surgery. Interestingly, however, repeated striatal D2R BP measurements during long-term follow-up (2 years) after RYGB surgery in 14 subjects of the original cohort reported by de Weijer et al. [45] demonstrated that the mean BMI declined and this decrease was associated with a significant increase in striatal D2R availability in this cohort [46]. These findings demonstrated that striatal D2R availability increases after long-term RYGB-induced weight loss and that the reduced D2R availability in obesity could be reversible. These findings support the idea that D2R downregulation could be a consequence of excessive eating and of obesity, rather than a cause, as was also reported in rats fed a highly palatable cafeteria diet [26,29]. However, taken altogether, the relationship between dopamine release or dopamine receptor availability and the obese phenotype remains unclear based on current brain imaging studies. The discrepancies among studies of D2R BP and BMI can be explained by use of different ligands across the different studies and individual variability in the experimental group, as well as different study designs with variability in metabolic status and diet types, measurement times, and so forth. Another question on the data from these studies relates to the real physiological relevance of D2R BP changes observed by brain imaging. It remains to be clarified whether the decrease in D2R availability represents a reduction of D2R density, which is hence associated with altered dopamine neurotransmission, or this decrease in D2R binding corresponds to an increase in the concentration of extracellular dopamine given that these ligands for brain imaging compete with endogenous dopamine for binding to the dopamine D2R. It has been postulated that decreased mesolimbic dopamine neurotransmission can induce a reward deficiency, leading to abnormal craving behaviors to compensate for this deficit [2,47]. Otherwise, increased mesolimbic dopamine release may be associated with impulsivity and is believed to be associated with increased incentive salience, which can result in excessive intake of highly palatable foods [29]. Future studies with new imaging technologies, which can provide direct and more precise central dopamine levels will be necessary to resolve this issue.
ROLE OF HYPOTHALAMIC DOPAMINE IN THE REGULATION OF FOOD INTAKE
Dopamine neurons and receptors in the hypothalamus
Beyond its role as part of the reward system from the mesolimbic brain area, dopamine can contribute to the regulation of normal appetite and food reward by interacting with hypothalamic homeostatic signals. In the hypothalamus, dopamine-containing neurons are present in the posterior hypothalamus and in the medial zona incerta (group All and A13), in the arcuate nucleus (group A12), and in the rostral periventricular region [47,48]. The dopamine neurons situated in the rostral part of the arcuate nuclei constitute tuberohypophysial dopamine neurons projecting to the neural and intermediate lobes of the pituitary [47,48]. The tuberoinfundibular (arcuato-infundibular) dopamine neurons, which are located in the arcuate nucleus and adjacent periventricular region, project to the external layer of the median eminence [47,48].
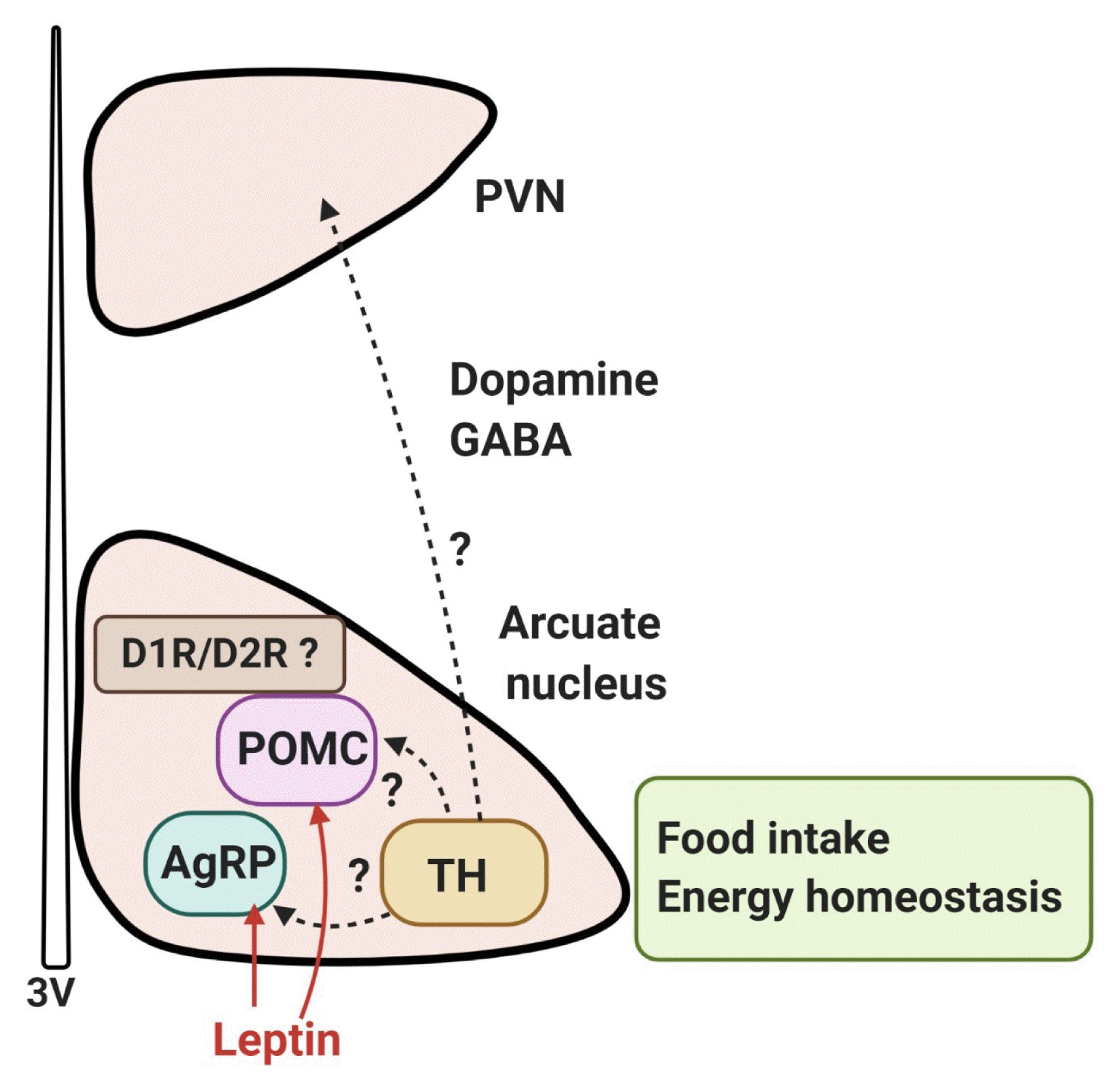
A systematic analysis of dopamine neurons in the hypothalamus revealed that in rodents, dopamine neurons (tyrosine-hydroxylase positive) are present throughout the hypothalamus, particularly in the arcuate nucleus and dorsomedial nucleus, with some areas of low density in the lateral hypothalamic area (LHA), except the zona incerta, where a high density of dopamine neurons is present [49]. Recently, it has been reported that dopamine neurons of the hypothalamic arcuate nucleus colocalize with vesicular GABA transporter and corelease GABA; these cells project to the hypothalamic paraventricular nucleus and communicate with other neurons, which suggests the possibility that they may regulate the excitability of synapsing neurons [49–51]. Furthermore, optogenetic stimulation of mouse arcuate dopamine neurons increased food intake; when these neurons were silenced, a long-term reduction in body weight occurred [51]. Optogenetic stimulation of these neurons inhibited pro-opiomelanocortin (POMC) neurons, possibly via D2R and dopamine excited neuropeptide Y (NPY)/agouti-related peptide (AgRP) neurons. Food deprivation also increased c-Fos expression and the activity of arcuate dopamine neurons, suggesting that hypothalamic dopamine neurons may directly contribute to the hypothalamic control of energy homeostasis by sensing energy-state-related signals and directly modulating the activity of other neurons within the hypothalamic feeding circuits [51]. These findings suggest that dopamine neurons in the hypothalamus can control food intake independently and/or by interacting with other neurons such as NPY/AgRP and POMC neurons to regulate energy homeostasis (Fig. 2).
These effects of hypothalamic dopamine signaling are mediated mostly by D1R and D2R; it has been reported that in the human brain there is a preferential expression of D2R as compared to D1R in the hypothalamus [52]. In the human brain, D2R mRNA-expressing cells have been found in most of the major hypothalamic nuclei, particularly the lateral and ventromedial nuclei (VMN) [53]. In rodents, expression of D2R is found in several hypothalamic regions, including the LHA, dorsomedial, paraventricular, VMN, and arcuate nuclei [54,55]. Cells expressing D1 receptor mRNA were detected in the paraventricular, supraoptic, and suprachiasmatic nuclei, but also observed to a lesser extent in the dorsal and medial nuclei, the VMN, and LHA [56].
Initial pharmacological studies using D1 and D2R agonists/antagonists showed that the modulation of hypothalamic dopamine receptor activity could regulate feeding behavior, although these studies had limitations in their ability to provide greater precision for cell-type or pathway-specific function in a behavioral response. Bina and Cincotta [57] reported that daily treatment of genetically obese, ob/ob mice with dopamine D1/D2R agonists for 2 weeks normalized body weight, hyperlipidemia, and hyperglycemia to the levels observed in lean mice and also reduced hyperphagia. Treatment with dopamine D1/D2R agonists also significantly reduced elevated levels of NPY and corticotropin-releasing hormone (CRH), which promote food intake, in the hypothalamus of obese mice. These findings suggest that dopaminergic D1/D2R coactivation may ameliorate the obesity phenotype in ob/ob mice, partially by modulating levels of both NPY and CRH [57].
Using a tumor-bearing anorexia rat model where meal size and number were decreased, Sato et al. [58] examined the role of D1 and D2R in the LHA and VMN, in association with feeding patterns (changes in meal number and size). Interestingly, hypothalamic expression of D1 and D2R mRNA was higher in anorectic tumor-bearing rats. Intra-VMN D1R antagonist injection in tumor-bearing rats decreased food intake with a decrease in meal size, while intra-VMN D2R antagonist administration in tumor-bearing rats increased food intake with an increase in meal number. In non-tumor-bearing free-feeding rats, food intake increased with an increase in meal size. In the LHA, there was no effect of D1R antagonist on food intake, whereas intra-LHA injection of a D2R antagonist in tumor-bearing and in non-tumor-bearing free-feeding rats increased food intake with an increase in meal number, suggesting that dopamine receptor subtypes in the hypothalamus are differentially involved in the regulation of food intake [58].
Recently, it has been shown that the D1R and D2R are located on POMC-immunopositive neurons, and co-localized with leptin receptors in the arcuate nucleus of rats and mice [59], raising the possibility that dopamine signals in the hypothalamus may be involved in the leptin receptor-mediated network (Fig. 2). Within the hypothalamic circuitry, it is probable that interactions between dopamine and leptin, a primary player in the homeostatic hypothalamic network of energy balance, will play a critical role. We will now briefly discuss the interaction of the hypothalamic dopamine system with the leptin network.
Interaction between the dopamine system and leptin
Leptin, an adipocyte-derived hormone, is a primary potent regulator of appetite and by binding to the leptin receptor in the hypothalamus, induces specific signaling cascades that suppress food intake [60,61]. It is well established that in the arcuate nucleus of the hypothalamus, leptin receptors are expressed in POMC neurons, which produce the anorectic peptide α-mela-nocyte–stimulating hormone [60–62]. On the other hand, leptin receptors are also expressed in neurons that produce the orexigenic peptides NPY and AgRP. Binding of leptin to leptin receptors in these neurons appears to regulate the activity of these neurons in opposite directions [60–62]. Early evidence has suggested interactions between leptin and the dopamine system. Initial findings of the expression of leptin receptors on dopamine neurons in the arcuate nucleus of the hypothalamus provided neuroanatomical evidence for the interactions between leptin and the dopamine system [62].
Kok et al. [63] reported a study about the acute effect of bromocriptine, a D2R agonist, on circadian leptin levels in obese women. Activation of D2R by bromocriptine treatment reduced circulating leptin levels in an obese woman, although the effect was modest (approximately 10% reduction compared with placebo), indicating that dopaminergic control through D2R may play a role in regulating leptin levels in humans [63]. Reciprocally, in a study of the effect of leptin on dopamine release from hypothalamic neuronal endings in vitro, leptin had no effect on basal release, but inhibited depolarization-induced dopamine release [64]. This suggests that dopamine enhances feeding in the hypothalamus, implying that the anorectic activity of leptin in the hypothalamus could be attributed to an inhibition of dopaminergic neuron firing.
Hagan et al. [65] examined the concentration of leptin in human plasma and cerebrospinal fluid (CSF), together with dopamine, and found that CSF dopamine levels were strongly correlated with plasma leptin levels. Both plasma leptin and CSF dopamine were positively associated independent of BMI, suggesting that dopaminergic systems are involved in the effects of leptin and that this interaction might be altered in obesity [65]. It is also possible that the amelioration of diabetes and obesity by dopamine agents involves leptin [65]. Clark et al. [66] investigated the effect of leptin administration via the intraperitoneal or intracerebroventricular route on hypothalamic dopamine levels in rats and observed that both intraperitoneal and intracerebroventricular administration of leptin treatment significantly decreased dopamine concentrations in the paraventricular nucleus, which is presumed to be associated with an opposite effect on thyrotropin hormone secretion exerted by dopamine and leptin [66,67]. However, it is also possible that this hypothalamic leptin-dopamine regulation can be relayed to other brain areas that influence motivated behaviors, which finally can be interfaced with the midbrain dopamine reward circuit. Our own work with dopamine D2R KO mice showed lower body weight and food intake in D2R KO mice than in wild-type littermates [68], and hypothalamic leptin signaling was enhanced in the absence of D2R, indicating that the hypothalamic dopamine receptor D2R-leptin interaction plays a role in the control of food intake and energy homeostasis [68].
The association between the dopamine receptor genotype and obesity in humans has been also investigated, demonstrating that genetic variants of the human obesity gene interact with DRD2, the gene that codes for D2R [69–72]. The most investigated polymorphism of DRD2 is allelic variants of the Taq1A polymorphism in the DRD2 gene, which is located within the protein-encoding region of a neighboring gene (ankyrin repeat and kinase domain containing 1 [ANKK1]) and it has been suggested that the Taq1A polymorphism affects D2R expression [70,71]. The Taq1A polymorphism has three allelic variants: A1/A1, A1/A2, and A2/A2; individuals with one or two copies of the A1 allele (i.e., A1/A1 and A1/A2 genotypes) have lower D2R density than those without an A1 allele (i.e., the A2/A2 genotype) [70,72].
Interestingly, an association of the DRD2 gene (the TaqI A1 allele) with the leptin receptor gene (LEPR) (Lys109Arg) was found in a sample of individuals with clinically severe obesity [73]. Individuals who carried one or more copies of TaqI A1 and were homozygous for the Lys109Arg LEPR variant had a significantly higher BMI than carriers of all other alleles, suggesting a possible critical interaction between dopamine and the leptin system through D2R, although no independent association between Lys109RArg and BMI was observed [73].
When D2R availability was examined using PET imaging in the human brain, shortly after RYGB surgery or after short-term caloric restriction in obese females with a very-low-calorie diet, body weight and dopamine D2R binding availability decreased, which was associated with a decrease in leptin concentrations [43,74]. It is possible that caloric restriction-induced decreases in leptin interact with the dopamine system, probably to comply with calorie restriction-induced energy homeostasis.
In a recent study, Lopez-Vicchi et al. [75] reported that female mice with a specific loss of lactotrope D2Rs (lacDrd2KO mice) that evokes hyperprolactinemia developed leptin insensitivity by 10 months of age, were overweight, showed increased food intake and body fat, and had higher serum leptin and insulin levels than the wild-type control group. In addition, these lacDrd2KO mice at 10 months of age showed increased Npy and Agrp gene expression in the arcuate nucleus and in the dorsomedial nucleus in the hypothalamus [75]. The obese lacDrd2KO mice displayed leptin resistance, as evidenced by blunted leptin response to food intake and altered leptin receptor signaling, specifically in the mediobasal hypothalamus [75]. These results suggest an important role of prolactin in metabolic regulation via hypothalamic regulation, but at the same time a role of D2R in lactotroph cells, which are under both hypothalamic dopamine release and feedback control to the hypothalamus.
CONCLUSIONS
Growing evidence connects the circuit controlling food intake, and recent findings have provide insights into specific populations of cells or circuits that control specific feeding-related behaviors and have helped demonstrate interactions between the homeostatic and reward circuits of feeding behavior. It is still clear that the dopamine reward circuit is the central regulator of motivation for food, although increasing evidence is emerging regarding the details of dopaminergic control in the homeostatic system in controlling eating behavior and obesity. Although these two systems appear to be functionally and anatomically distinct, one system (e.g., the hypothalamic dopamine system in control of food intake and energy homeostasis) may provide a substantial role of the dopamine system over integrative neural regulation of food intake in association with another system, the mesolimbic dopamine system. Therefore, one can argue that these distinct dopaminergic circuits for control of feeding behavior are closely connected through a network that most likely employs various intermediates, such as leptin, insulin, and other appetite-controlling peptide hormone and neurotransmitters with specific circuits. Current and upcoming breakthroughs in cell type- and circuit-specific genetic manipulation and cellular imaging tools will facilitate an improved understanding of the converging circuitry of control of food intake by linking energy state-associated signals to reward-related behaviors.
 XML Download
XML Download