

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Adipocyte mitochondria are essential organelles for maintaining whole-body metabolism in rodents and humans. Mitochondrial dysfunction in adipocytes has been reported in patients with obesity, insulin resistance, and type 2 diabetes [1]. Although compromised mitochondrial function in adipose tissue may arise from obesity and diabetes, it remains uncertain whether this is a consequence of, or a primary contributor to, the development of insulin resistance. Furthermore, we have a limited understanding of the impact of primary mitochondrial stress in adipose tissue, and its possible effects on systemic energy homeostasis. Mitochondrial proteins are comprised of mitochondrial DNA (mtDNA)-encoded and nucleus-encoded proteins, and around 99% of mitochondrial proteins are encoded by the nucleus [2]. Thus, the maintenance of mitonuclear protein balance is important for normal cellular function. Defective mitochondria communicate with the nucleus through retrograde signaling, known as mitonuclear communication, to maintain proper mitochondrial function and organismal homeostasis [3]. Past and recent studies have shown that mitochondrial stress activates the mitochondrial unfolded protein response (UPRmt), which is mediated by cell autonomous and cell non-autonomous pathways [4,5].
Mitochondrial protein homeostasis (proteostasis), which is regulated by chaperones and intrinsic proteases, acts as a key mechanism of mitochondrial quality control [6]. In terms of metabolism, key proteases related to the UPRmt, such as caseinolytic peptidase P (ClpP) and LON protease (LONP1), have been reported to affect systemic insulin sensitivity and glucose metabolism, respectively [7,8]. In addition to the effects of mitochondrial proteases, knockout (KO) models studying the effects of mitochondrial ribosomal defects have also shown compromised proteostasis and altered systemic metabolism.
Crif1 is a component of the large mitochondrial ribosomal subunit, and its deficiency results in abnormal proteostasis in the mitochondrial matrix of mouse embryonic fibroblasts [9]. In skeletal muscle-specific Crif1 KO mice, oxidative phosphorylation (OxPhos) function and the oxygen consumption rate decreased, but systemic energy homeostasis was maintained by UPRmt induction and metabokine production [10]. However, despite studies showing that adipocyte mitochondrial function and quality control are closely related to systemic energy metabolism, there are not many studies on causes of adipocyte-specific OxPhos dysfunction and the effects of metabokines on whole-body metabolism.
Adipose tissue inflammation is considered a major contributor to systemic insulin resistance by inducing pro-inflammatory cytokines secreted from macrophages and cytotoxic T cells [11]. Adipose tissue macrophages (ATMs) can be classified into two groups: classically (M1-like) and alternatively (M2-like)–activated macrophages. Increased macrophage number is characteristic of obese mice and humans [12]. In particular, M1 macrophages, which produce pro-inflammatory cytokines, were found to be elevated in obese individuals. M1 macrophages are characterized by high levels of glycolysis and inducible nitric oxide synthase expression, and secrete tumor necrosis factor alpha and interferon gamma (IFNγ) [13]. By contrast, lean individuals had elevated levels of M2 macrophages, which are characterized by high levels of OxPhos, fatty acid oxidation, arginase 1 expression, and secretion of the anti-inflammatory cytokine interleukin 10 (IL-10) [14]. The intact mitochondrial oxidative metabolism of M2 macrophages is a prerequisite for this anti-inflammatory phenotype [15]. Therefore, inhibition of oxidative metabolism impairs polarization to the M2-like phenotype, but shifts macrophages towards the M1-like state and leads to obesity, inflammation, and insulin resistance [16]. However, it is unknown whether treatments that can improve the oxidative function of macrophages can reverse insulin resistance and adipose inflammation. In this review, we discuss whether the UPRmt and metabokine production caused by lower OxPhos in adipocytes and ATMs regulate systemic energy metabolism and glucose homeostasis.
THE MITOCHONDRIAL UNFOLDED PROTEIN RESPONSE IN ADIPOCYTES IS LINKED TO INCREASED ENERGY EXPENDITURE AND PROTECTION AGAINST DIET-INDUCED OBESITY
The mitochondria are essential for adipose tissue function, including adipogenesis [17], lipolysis [18], and fatty acid re-esterification [19], which ultimately balances whole body homeostasis. Metabolic challenges such as nutrient excess, aging, and excess free fatty acids lead to mitochondrial dysfunction through the production of mitochondrial reactive oxygen species (ROS) [20]. These lead to reduced mitochondrial biogenesis [21], aggravated inflammation [22], and subsequently promote changes in energy homeostasis and insulin sensitivity [23].
Studies in ob/ob and diabetic mice have shown compromised mitochondrial function in white adipose tissue (WAT), including decreased mitochondrial number, mtDNA quantity, and electron transport chain enzymatic activity [24,25]. This is mirrored in human patients presenting with obesity, insulin resistance, and type 2 diabetes, in whom mitochondrial function-related genes, adenosine triphosphate production, and oxygen consumption are decreased in adipose tissue [26]. A commonality in these mitochondrial stress environments is increased production of ROS, which alters adipogenesis and lipolysis in adipocytes in both mice and humans [27,28]. Furthermore, profound adipose tissue mitochondrial dysfunction can lead to lipodystrophy with insulin resistance and hepatic fat accumulation in mice [29]. These studies reveal that adipocyte mitochondrial dysfunction is intricately involved in the onset of systemic insulin resistance.
Nonetheless, several studies have questioned the linear relationship between mitochondrial and metabolic dysfunction. A study in adipocyte-specific Pgc1β-deficient mice demonstrated that decreased mitochondrial oxidative capacity in adipocytes was not sufficient for insulin resistance [30]. In addition, Vernochet et al. [31] reported that despite a lower mtDNA quantity in the adipocytes of mitochondrial transcription factor A KO mice, the KO mice had higher energy expenditure (EE) and were protected from high fat diet (HFD)-induced obesity and insulin resistance. Further, Bhaskaran et al. [7] demonstrated that the loss of ClpP, which is a mitochondrial quality control protease, protected mice from diet-induced obesity with increased EE and insulin sensitivity. Although a number of investigations have investigated the association between mitochondrial function and systemic insulin sensitivity (Table 1) [30–34], the causes or effects of mitochondrial function in adipocyte and systemic metabolism remain controversial.
Several past and recent studies have discovered that mitochondrial dysfunction causes cellular stress and subsequent metabokine secretion in various species [5,10,35–37]. The UPRmt, which is conserved from worms to mammals [38], has beneficial effects on whole-body metabolism through cell autonomous and cell non-autonomous pathways [7,10,39]. The cell non-autonomous component of the UPRmt is mediated by secretory proteins including metabokines, which affect distal organs and requires further mechanistic insight [5]. However, most studies of this phenomenon have used lower organisms [40,41], global KO mouse models [42], or transgenic mouse models [43,44]; thus, the effects of this response in specific tissues remain incompletely understood.
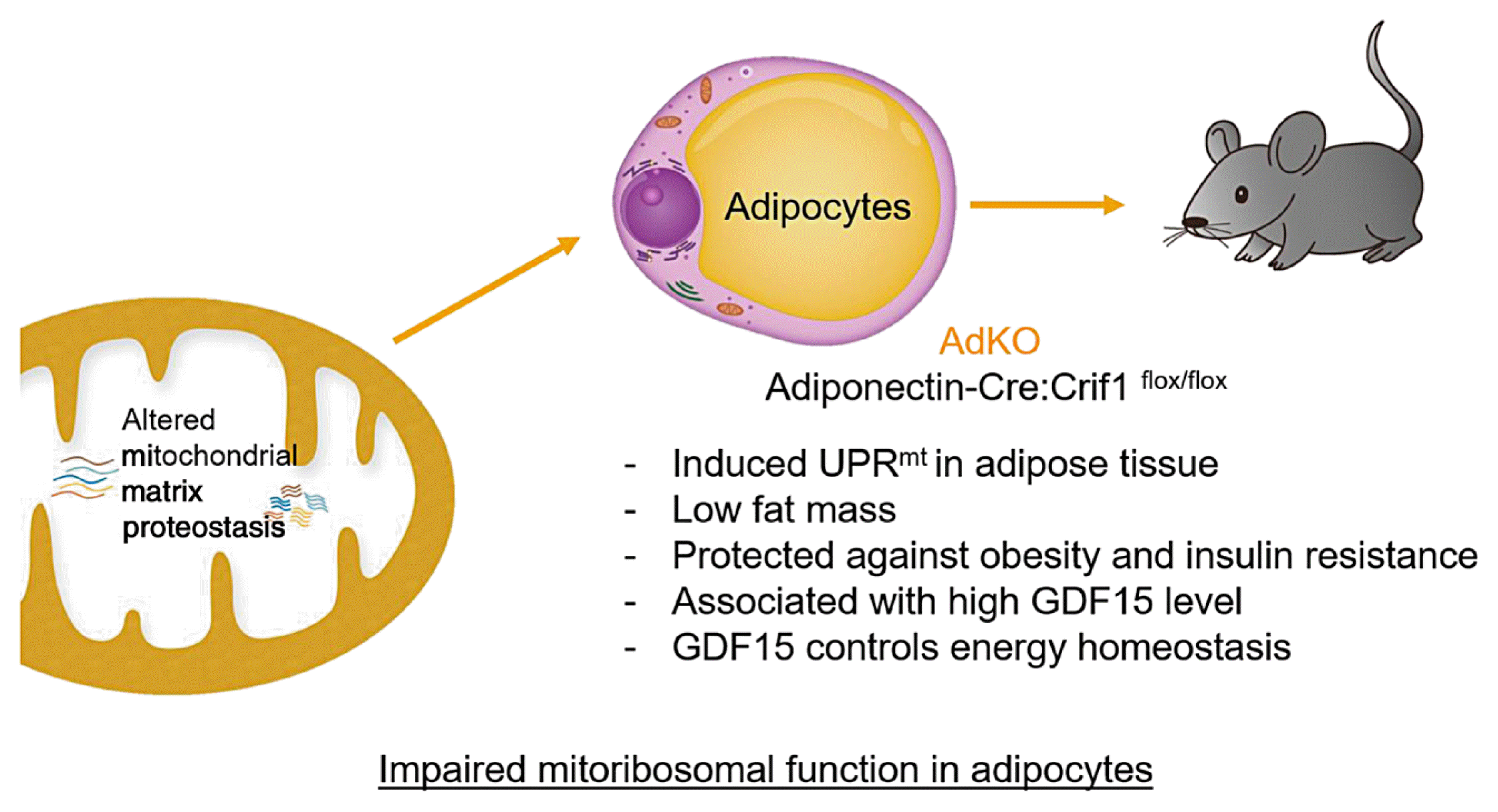
In particular, adipose tissue is a pivotal organ that plays a role in maintaining whole-body homeostasis through energy storage and adipokine secretion [45]. We demonstrated that adipocyte-specific activation of the local UPRmt was protective against the metabolic defects of diet-induced obesity in mice with adipocyte-specific deletion of the Crif1 gene (AdKO) [32]. AdKO mice fed an HFD showed improved metabolic phenotype, including decreased fat mass and fatty liver, and increased EE with uncoupling protein 1 (UCP1) induction. The UPRmt was induced in adipocytes, and through RNA sequencing analysis, growth differentiation factor 15 (GDF15) was observed to increase dramatically in AdKO mice. Double KO of GDF15 and adipose-specific Crif1 abrogated the beneficial metabolic effects seen in AdKO mice, with increased weight gain and decreased EE and UCP1 expression (Fig. 1). This indicates that GDF15 modulates body weight and energy homeostasis in AdKO mice. Studies on global Gdf15 KO have confirmed that GDF15 has a protective effect on obesity [42], but further studies on the mechanisms of UPRmt and metabokine secretion are needed. While the evidence is only suggestive for the role of UPRmt in metabolic disease in mammals, metabokines such as GDF15 will serve as a useful diagnostic biomarker for human mitochondrial disease, as well as a potential therapeutic modality for metabolic diseases.
IMPAIRED MITOCHONDRIAL OXIDATIVE FUNCTION IN ADIPOSE TISSUE MACROPHAGES RESULTS IN ADIPOSE TISSUE INFLAMMATION AND INSULIN RESISTANCE
Recent investigations have demonstrated that immune cells residing in adipose tissue are involved in the regulation of systemic metabolic homeostasis [46]. Inflammation induced by immune cell infiltration in adipose tissue is a feature of adipose tissue dysfunction. Among the various immune cells, ATMs play a critical role in the adipose tissue microenvironment by determining anti- and pro-inflammatory responses. In obese mice and humans, macrophage infiltration is increased, particularly of M1-polarized macrophages, which are known to secrete pro-inflammatory cytokines and induce inflammation and systemic insulin resistance [47–49]. Mitochondrial function has been shown to play an important role in macrophage polarization.
Several studies on macrophage mitochondria have been conducted in vitro and in vivo. M1-polarized macrophages induced by lipopolysaccharide and IFNγ have low mitochondrial respiration with decreased dependence on the Krebs cycle [50]. On the contrary, M2-polarized macrophages have an intact Krebs cycle, OxPhos, and increased fatty acid oxidation, which is required for the anti-inflammatory response [51]. Studies on the M1-to-M2 macrophage shift have suggested that it is possible to halt the progression of chronic inflammation in adipose tissue [52,53]. Although the impaired oxidative function of pro-inflammatory M1 macrophages affecting adipose tissue inflammation and systemic insulin resistance has been extensively studied, it remains unclear whether primary OxPhos deficiency in macrophages causes insulin resistance associated with adipose tissue inflammation.
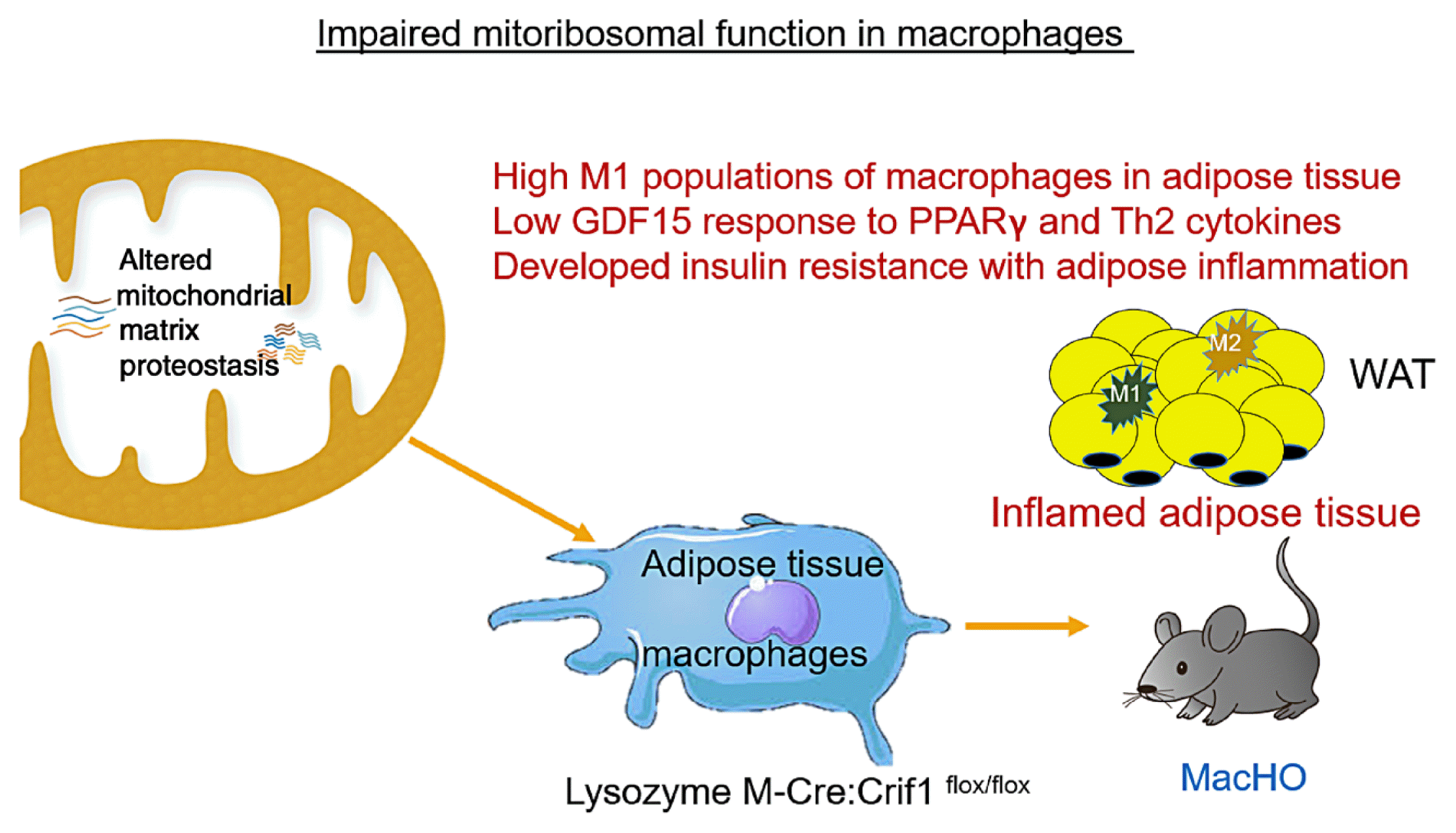
We demonstrated that a myeloid cell-specific mitoribosomal defect in LysM-Cre mice presented an increased M1-polarized macrophage state in adipose tissue, and the expression of common M2 macrophage genes such as Arg1, Ym1, and GDF15 expression were reduced [54]. Additionally, we showed that reduced mitochondrial oxidative function in macrophages precipitated adipose inflammation, including increased M1 macrophages, and systemic insulin resistance in HFD-fed mice (Fig. 2). It is known that peroxisome proliferator-activated receptor gamma (PPARγ) and signal transducer and activator of transcription 6 activate M2 macrophages by increasing oxidative metabolism and mitochondrial biogenesis [55,56]. M2 macrophages have been linked to an anti-inflammatory response and results in secretion of IL-10, transforming growth factor-β1, Ym1, and Fizz1. These proteins are paracrine factors that regulate the adipose tissue environment, and are involved in the determination of the polarization of ATMs. We observed that GDF15 was increased by the Th2 cytokine IL-4 and PPARγ agonist rosiglitazone.
In macrophages, GDF15 treatment results in increased mitochondrial respiration and increased expression of genes related with fatty acid oxidation. By observing the increase in respiration, palmitate oxidation, and fatty acid oxidation-related gene expression in macrophages treated with GDF15, it can be seen that GDF15 induces the shift of the macrophage phenotype by improving oxidative function. Furthermore, it was observed that M2 polarization was not induced in macrophages from GDF15 KO mice, even with IL-4 treatment, and glucose tolerance was impaired by adoptive transfer of bone marrow-derived macrophages from GDF15 KO mice. These results support the role of GDF15 as a regulator of M2 macrophage polarization, indicating that GDF15 is a protein that protects against WAT inflammation. The beneficial effects of GDF15 were further demonstrated by several studies. Upregulated GDF15 has a protective role in advanced atherosclerosis, macrophage accumulation, and apolipoprotein E-deficient mice [57,58]. Moreover, GDF15 transgenic mice showed resistance to diet-induced obesity and increased insulin sensitivity with lower NLR family pyrin domain containing 3 (NLRP3) inflammasome activity in adipose tissue [59], verifying the role of GDF15 in controlling macrophage-mediated adipose inflammation.
Collectively, GDF15 can modulate the adipose tissue immune environment by increasing the oxidative function of macrophages and shifting the polarization to M2-like macrophages. Thus, it can be concluded that GDF15 has both an autocrine and paracrine effect, which can improve systemic metabolic homeostasis.
THE METABOKINE GDF15 COORDINATES CELLULAR AND INTERCELLULAR HOMEOSTASIS IN ADIPOSE TISSUE
The concept of a mitokine was initially suggested in a Caenorhabditis elegans model, where mitochondrial dysfunction in the brain triggered UPRmt activation non-autonomously in gut cells, and was mediated by an unknown factor that was termed a mitokine [5]. It is known that mitokines are secreted from tissues with primary mitochondrial dysfunction, but in the complex mammalian system, a mitokine can be defined as a metabokine capable of controlling systemic metabolism by peripheral and central nervous system pathways [60,61].
While FGF21 is also considered as a major metabokine, it has already been discussed extensively by Geng et al. [60], and the present article will focus on the metabokine GDF15. GDF15 has been studied for its role in models of cancer [62], aging [63], longevity [64], and mitochondrial dysfunction [65]. The expression of GDF15 was observed in mouse and human adipose tissue, and was found to be secreted from adipocytes, suggesting a paracrine role in the modulation of adipose tissue function and body fat mass [66]. We screened possible metabokine candidates in the adipose tissue of AdKO mice, and showed that metabokines play an important role in the phenotype associated with OxPhos dysfunction in AdKO mouse adipocytes [32]. The anti-inflammatory effect of GDF15 was extensively studied and discussed in the previous section [67,68]. However, the mechanism by which metabokines regulate systemic metabolism in tissue-specific mitochondrial dysfunction models is not fully understood.
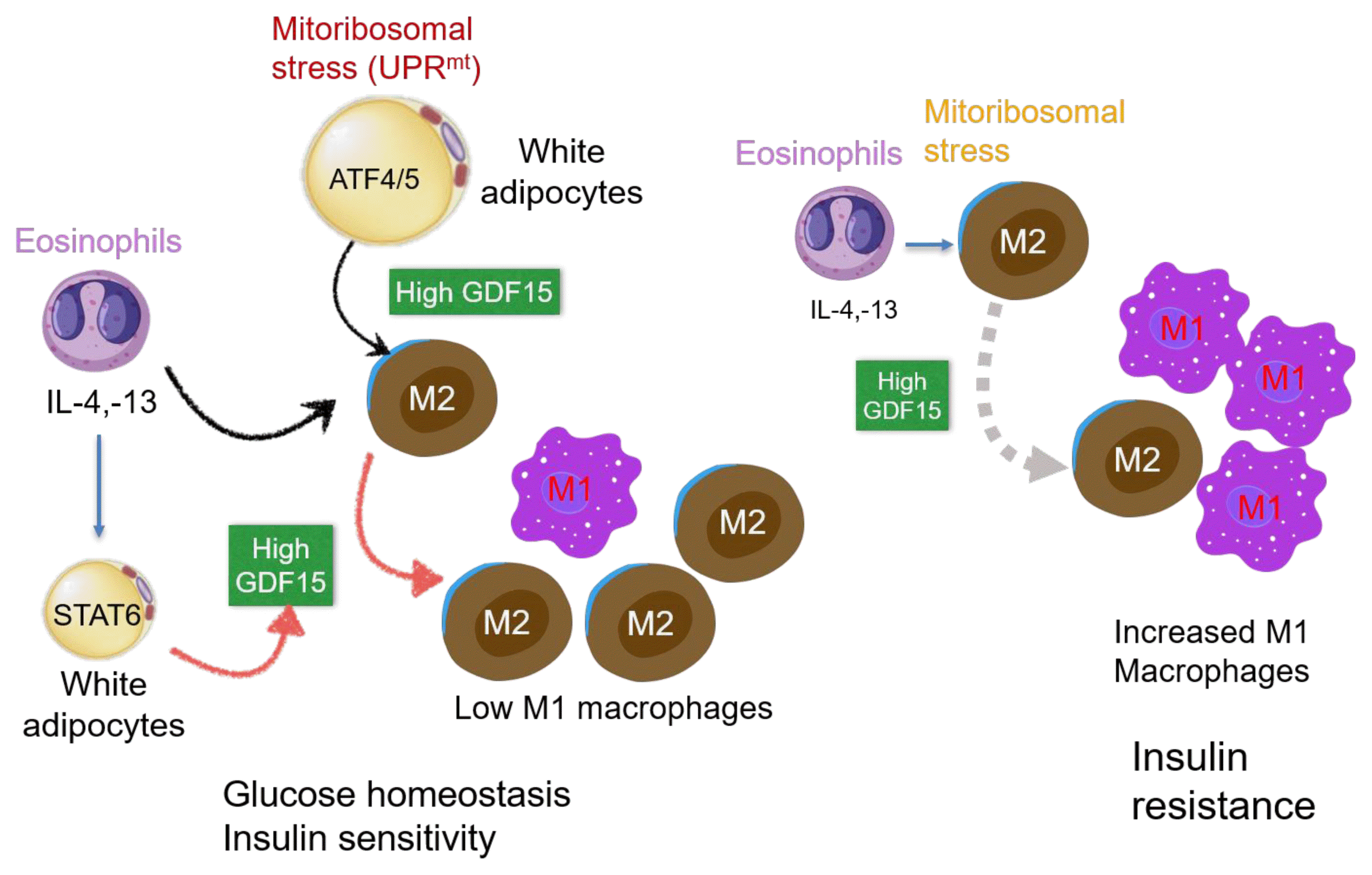
We recently demonstrated that AdKO mice with whole-body GDF15 deletion have increased M1 macrophage proportions and reduced M2 macrophage expression in adipose tissue. Furthermore, we observed that myeloid-specific mitoribosomal defects induced M1 macrophage polarization and decreased M2 macrophages in adipose tissue [54]. It was also observed that M2 macrophage polarization by IL-4 treatment did not occur in macrophages of GDF15 KO mice, suggesting that GDF15 is a regulator of anti-inflammatory macrophage polarization, while also acting as a protective metabokine against WAT inflammation [54]. Adipose tissue inflammation is associated with dysregulated immune cell function and a high M1/M2 macrophage ratio, and is involved in systemic insulin resistance [69,70]. M2 macrophages are associated with Th2 cytokines, which are secreted from eosinophils [71]. Th2 cells in adipose tissue suppress inflammatory responses by secreting the anti-inflammatory cytokine IL-10 [72]. IL-4 and IL-13, secreted from eosinophils in adipose tissue, are associated with macrophage reconstitution and are involved in improved glucose tolerance and insulin sensitivity [71]. We observed that GDF15 was required for IL-13-induced M2 macrophage polarization via the Janus kinase (JAK)-signal transducer and activator of transcription 6 (STAT6) pathway. It was shown that administration of IL-13 to wild-type mice fed an HFD improved glucose tolerance, which was not observed in GDF15 KO mice. These findings suggest that the improvement of systemic metabolism by IL-13, a Th2 cytokine, is mediated by GDF15 and acts as a regulator of anti-inflammation and systemic glucose homeostasis (Fig. 3) [73].
The metabolic action of GDF15 was shown to regulate food intake and body weight through binding with GDNF family receptor alpha like (GFRAL), which is localized in the area postrema and the nucleus tractus solitarius region of the hindbrain [74–76]. Weight loss and food intake reduction were eliminated in GFRAL KO mice, suggesting that GDF15 has a central role in appetite regulation [74–76]. However, other studies suggested that GDF15 may have a peripheral action [77,78]. Thus, further studies of the peripheral receptor of GDF15 are necessary to identify its peripheral metabolic activity. Collectively, GDF15, is a metabokine that is increased by the mitochondrial stress response, and possibly plays a peripheral role in regulating intercellular homeostasis through not only central nervous system-mediated action via GFRAL, but also through immune and metabolic reprogramming through undiscovered receptors (Fig. 4).
CONCLUSIONS
GDF15 is highly increased in various pathologies including obesity, cancer, and mitochondrial stress. Whole-body KO of Gdf15 in mice showed increased weight gain due to higher food intake. An adipocyte-specific mitoribosomal defect mouse model (AdKO) showed dual activation of cell-autonomous (chaperones and proteases) and cell non-autonomous metabokine mechanisms in WAT. This finding implies a novel role for the UPRmt and metabokine secretion in adipose tissue homeostasis, which can regulate both systemic glucose homeostasis and EE as part of an organismal adaptation to local mitochondrial stress. We have shown that adipose OxPhos function from WAT can influence systemic glucose homeostasis and EE in pathologic states such as diet-induced obesity via the metabokine GDF15. Several studies have discovered that GDF15 plays a role in appetite regulation and body weight gain through its binding with GFRAL, but it is not known which receptors and mechanisms are involved to exert the peripheral effects of GDF15. Thus, further research is needed to discover the mode of action of GDF15 in peripheral tissues.

 XML Download
XML Download