

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Glucagon-like peptide-1 (GLP-1) is secreted from gastrointestinal endocrine L-cells upon food intake. The cellular density of L-cells gradually increases along the length of the small intestine, with the lowest density in the duodenum and the highest in the distal ileum [1]. Therefore, as more unabsorbed nutrients reach further down the distal small intestine, more GLP-1 is secreted. Excess food intake or rapid gastric emptying that exceeds the absorptive capacity of the upper small intestine increases the amount of unabsorbed nutrients in the lumen of the more distally located small intestine, thereby increasing the amount of GLP-1 secretion. Postprandial production of GLP-1 increases insulin secretion from pancreatic beta cells and suppresses glucagon secretion from pancreatic alpha cells. Teleologically, increased GLP-1 secretion plays a role as an ileal brake to maintain “a normal speed” of energy metabolism by decelerating gastric emptying and suppressing food intake. The physiological roles of GLP-1 are more than sufficient for it to be targeted for the development of anti-diabetic and anti-obesity medication [2].
Go to : 
EVOLUTION OF GLP-1 THERAPY
In 1983, GLP-1 was found in the quest for the discovery of a glucagon gene [3]. Biologically active GLP-1 consists of 30 or 31 amino acids (GLP-1 (7–36) amide or GLP-1 (7–37), respectively), which are derived from the proglucagon peptide. Despite the pleiotropic properties of GLP-1, including its insulinotropic effect found in rats, pigs, and humans, its extremely short plasma half-life of less than 2 minutes constituted a huge brick wall in the path of GLP-1 to be developed as a glucose-lowering medication. Later, dipeptidyl peptidase-4 (DPP-4) was reported to rapidly remove the N-terminal diamino acid peptide of GLP-1 by recognizing alanine and proline at the penultimate position [4].
In 1992, a proof-of-concept study first suggested the potential of GLP-1 as an anti-diabetes drug, as a 3-hour continuous intravenous GLP-1 infusion significantly lowered the meal-related insulin requirement in patients with diabetes; decreased postprandial glucose levels, and increased the insulinogenic index in normal subjects [5]. In 2002, a 6-week subcutaneous infusion of GLP-1 showed a robust glucose-lowering effect in patients with type 2 diabetes [6].
Henceforth, researchers devised numerous strategies for drug development, mainly by evading the enzymatic degradation of bioactive GLP-1 peptide by DPP-4. Of note, exendin-4 was serendipitously discovered in the saliva of the Gila monster (Heloderma suspectum) and was revealed to be a GLP-1 analog that is resistant to DPP-4 action in 1992, even before the mechanism of GLP-1 degradation was identified [7]. More than a decade later, in 2005, exenatide (a synthetic version of exendin-4) was approved as the first anti-diabetes medication in the GLP-1 receptor agonist class by the U.S. Food and Drug Administration. This small peptide is eliminated by the kidneys and has a plasma half-life of approximately 3 hours, requiring twice-daily subcutaneous injections. The current approved dose of exenatide is 5 or 10 μg given twice-daily. A modified version of exenatide was developed by adding lysine residues to the C-terminal (lixisenatide). Although it displays a very similar pharmacological profile to that of exenatide, lixisenatide is given once-daily with a dose of 10 or 20 μg. To increase the plasma half-life, a slower-releasing formula of exenatide was developed by incorporating to a polymer microsphere, permitting a once-weekly injection. More recently, a long-acting exendin-4-based analog bound to the Fc portion of immunoglobulin G (IgG) known as efpeglenatide is under clinical development, and would also be suitable for a once-weekly injection [8,9].
Analogs based on the human GLP-1 structure have also been developed [10,11]. To avoid enzymatic cleavage by DPP-4, the N-terminal penultimate amino acid has been replaced with other amino acids (albiglutide, dulaglutide, and semaglutide) or added with a fatty acid moiety (liraglutide and semaglutide), so that albumin binds and shields the enzymatic attack site. To increase the plasma half-life by avoiding renal elimination, GLP-1 analogs were modified by adding acyl groups (liraglutide and semaglutide) or large molecules such as albumin (albiglutide) and the Fc portion of IgG (dulaglutide).
Go to :
NOVEL DELIVERY ROUTES FOR GLP-1
Despite the considerable momentum to acclaim GLP-1 as a preferred first-line injection therapy for the treatment of type 2 diabetes, other routes of administration than injections have long been pursued. Firstly, a GLP-1 tablet was developed to be absorbed through the oral buccal mucosa [12]. However, this pioneering approach was futile due to the relatively thick mucosal barrier and continuous flow of saliva. Secondly, inhaled GLP-1 was developed, with the goal of facilitating absorption through the alveolar epithelium [13]. The large surface area of the pulmonary bed enables rapid absorption of GLP-1, which can mimic postprandial GLP-1 secretion. Indeed, inhaled GLP-1/technosphere reaches peak plasma GLP-1 concentration within 5 minutes after inhalation in humans. Although this is an attractive approach that spurred great interest, caution should be exercised as standard dosing may be a challenge depending on individual inhalation technique. The long-term safety of inhaled GLP-1 also needs to be established since GLP-1 receptors are abundantly expressed in pulmonary tissue. Thirdly, implantable devices could be an option to replace injections. ITCA650 is a mini-osmotic pump that continuously releases exenatide into subcutaneous tissue, thereby mimicking the effects of a long-acting GLP-1 receptor agonist, and is free from compliance issues [14]. Of interest, a magnetically actuated implantable pump was proposed as a means to deliver exenatide to subcutaneous tissue on demand before a meal, thereby recapitulating postprandial GLP-1 secretion [15]. The magnetically actuated implantable pump produced promising results with efficacy comparable to that of subcutaneous injections in animal models, underscoring its potential as a viable therapeutic option in humans. Lastly but not least, oral GLP-1 receptor agonists would be the most favorable and acceptable option as a substitute for injection therapy, as discussed below.
Go to :
ORAL SEMAGLUTIDE
Oral semaglutide is the first noninjectable formulation of a GLP-1 receptor agonist approved by the U.S. Food and Drug Administration. While enteral administration offers several advantages over other routes of drug delivery in terms of convenience, promoting medication compliance, it faces several obstacles, of which the most widely recognized are inefficient absorption owing to physical barriers and an acidic microenvironment, along with degradation by proteases and the first-pass metabolism in the liver. Oral semaglutide overcame the major impediments of oral peptide delivery by being co-formulated with the permeation enhancer sodium N-[8-(2-hydroxybenzoyl) amino] caprylate (SNAC). SNAC is an acylated amino acid designed to improve the oral bioavailability of parenteral drugs that was extensively tested on cyanocobalamin and macromolecular drugs such as heparin and insulin [16,17].
Oral semaglutide has an advantage over oral insulin in that a fixed dosage can be safely administered with minimal hypoglycemic risk. Oral semaglutide incorporates SNAC, which facilitates transcellular absorption via the gastric mucosa through various mechanisms including increased lipophilicity, transforming semaglutide to a more permeable monomeric form, and local elevation of pH and the resultant pepsin-inhibiting effect [18]. The mean oral bioavailability of semaglutide reached 0.4% to 1.0% when co-formulated with 300 mg of SNAC in humans [19].
In the dose-finding phase 2 trial, the hemoglobin A1c (HbA1c) reduction from baseline to 26 weeks was −0.7% to −1.9% with varying doses of oral semaglutide (2.5 to 40 mg once-daily), −0.3% with placebo, and −1.9% with 1 mg of once-weekly subcutaneous semaglutide [20]. A series of 10 phase 3 multicenter, randomized clinical studies as part of the Peptide InnOvatioN for Early diabetes tReatment (PIONEER) program were conducted to compare the efficacy and safety of oral semaglutide with either placebo or active comparators [21–31]. Of note, a gradual dosage increment to 14-mg once-daily oral semaglutide treatment showed superiority in reducing HbA1c compared to 25 mg of empagliflozin (PIONEER 2: baseline HbA1c 8.1%, −1.3% vs. −0.9% for oral semaglutide to active comparator, respectively), 100 mg of sitagliptin (PIONEER 3: baseline HbA1c 8.3%, −1.1% vs. −0.7%), 1.8 mg of liraglutide (PIONEER 4: baseline HbA1c 8.0%, −1.2% vs. −0.9%), and 0.75 mg of dulaglutide (PIONEER 10: baseline HbA1c 8.3%, −1.7% vs. −1.4%).
According to a meta-analysis, oral semaglutide (14 mg once-daily) resulted in a sustained weight loss compared to placebo (−2.99 kg; 95% confidence interval [CI], −3.69 to −2.30) [32]. This weight reduction effect, one of the salient merits of GLP-1 receptor agonists, appeared slightly inferior to that of weekly subcutaneous semaglutide (−4.11 kg; 95% CI, −4.85 to −3.37) [33]. Since higher-dose formulations of oral and subcutaneous semaglutide are under development, it needs to be seen if they will be approved as anti-obesity drugs [34].
Cardiovascular safety has been evaluated with both oral and subcutaneous semaglutide. Oral semaglutide exhibited a neutral effect on major adverse cardiovascular events (hazard ratio [HR], 0.79; 95% CI, 0.57 to 1.11) in PIONEER 6, which was performed with 3,183 patients (baseline HbA1c, 8.2%; established cardiovascular disease [CVD], 84.7%; mean duration of type 2 diabetes, 14.9 years); intriguingly, although oral semaglutide did not prove superiority over placebo for the primary outcome, it significantly lowered the incidence of death from cardiovascular causes (HR, 0.49; 95% CI, 0.27 to 0.92). In Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN-6) trial involving 3,297 patients (baseline HbA1c, 8.7%; established CVD, 83.0%; mean duration of type 2 diabetes, 13.9 years), 0.5 or 1.0 mg of once-weekly subcutaneous semaglutide achieved superiority over placebo in reducing major cardiovascular adverse events (HR, 0.74; 95% CI, 0.58 to 0.95). It is notable that the absolute value of the HR was in fact similar to that in the SUSTAIN-6 trial. Since the follow-up duration of the two studies were different (15.9 months vs. 25.2 months for the PIONEER 6 and SUSTAIN-6 trials, respectively) [35,36], the HR of the PIONEER 6 study may have been statistically significant if the follow-up duration had been longer. A phase 3 clinical study with a higher dosage of daily 25 and 50 mg of oral semaglutide is about to be initiated, in the hope of obtaining promising data for both body weight reduction and cardiovascular benefits [34,37].
Consistent with other GLP-1 receptor agonists administered through subcutaneous injections, the most frequently recorded adverse events of oral semaglutide treatment were nausea and vomiting; a gradual dosage increment to 14-mg once-daily oral semaglutide treatment had gastrointestinal side effects parallel to those of 1 mg of once-weekly subcutaneous semaglutide [32,33]. To optimize effective absorption, patients are required to adhere to complicated dosing conditions of taking the drug in a fasting state (at least 30 minutes before any food intake) with no more than 4 ounces (approximately 120 mL) of plain water. Therefore, issues exist with drug compliance, and the substantial food-drug interactions may potentially generate inconsistent effects in the real-world setting.
Go to :
SMALL MOLECULE ORAL GLP-1 RECEPTOR AGONISTS IN DEVELOPMENT
As an alternative approach, non-peptide, small molecule oral GLP-1 receptor agonists offer a promising option. Earlier ambitious but futile attempts to develop these non-peptide drugs have been reviewed in detail elsewhere [10,38]. For many years, this alluring perspective seemed inconceivable; a comprehensive exploration of the structure-activity relationship of GLP-1 receptor activation in response to natural GLP-1 (7–36) amide revealed an extensive interaction of the C-terminus of GLP-1 with the peptide-binding groove of the N-terminal extracellular domain (ECD) of the GLP-1 receptor. The subsequent conformational transition causes the N terminus of GLP-1 to bind to the deep transmembrane (TM) domain pocket, allowing assembly of the downstream transformation of TM helices to enable G protein coupling. The key insulinotropic actions of GLP-1 via the class B1 G-protein-coupled receptor (GPCR) are exerted by cyclic adenosine monophosphate (cAMP) formation, which, in conjunction with increased Ca2+ levels, promotes exocytosis of insulin-containing vesicles [39]. Mimicking the initial multiple, extensive interactions with the ECD seemed implausible to achieve with smaller, non-peptide molecules.
Nevertheless, the search for orally administrable compounds that retain the impressive properties of GLP-1 receptor agonists persisted (Table 1). An analysis of the GLP-1 receptor revealed not only the aforementioned molecular details related to the interactions, but also the prerequisite amino acid residue that explained the species selectivity in binding of non-peptide molecules [40,41]. Specifically, while the GLP-1 peptide sequence is identical across mammalian species, the human GLP-1 receptor differs from that of the rat GLP-1 receptor at 42 amino acid residues. Of those, tryptophan at position 33 in the ECD (Trp33ECD) confers species selectivity; subsequent preclinical studies have thus screened agonists with primates with Trp33ECD or receptor mutants with substitution of Ser33ECD by Trp33ECD.
Table 1
Comparison of Peptide and Small Molecule GLP-1 Receptor Agonists
| Name | Peptides | Small molecules | |||
|---|---|---|---|---|---|
|
|
|
||||
| GLP-1 (7–36) | Semaglutide | TT-OAD2 | LY3502970 | PF-06882961 | |
| Molecular formula | C149H226N40O45 | C187H291N45O59 | C50H49Cl4N3O6 | C48H48F2N10O5 | C31H30FN5O4 |
|
|
|||||
| Molecular weight | 3,297.6 g/moL | 4,113.6 g/moL | 929.7 g/moL | 883.0 g/moL | 555.6 g/moL |
|
|
|||||
| Trp33 residue | Not necessary | Not necessary | Not necessary | Necessary | Necessary |
|
|
|||||
| cAMP/PKA pathway | Full agonist | Full agonist | Partial agonist | Partial agonist | Full agonist |
|
|
|||||
| Beta-arrestin pathway | Full agonist | Full agonist | No effect | No effect | Partial agonist |
|
|
|||||
| Oral dosage | NA | 3, 7, 14 mg | NA | 0.05, 0.15, 0.45 mg/kga | 3–300 mg |
|
|
|||||
| Oral bioavailability | Negligible | 0.4%–1% | NA | 21%–28%a | Dose-dependentb |
![]()
Two of the few recently recognized small molecules that succeeded in entering clinical development, namely LY3502790 and PF-06882961, activate the canonical G protein signaling activity only in the GLP-1 receptor with Trp33ECD [38,42]. This was a phenomenal discovery, since primate-specific Trp33ECD served as a critical point in the binding of small molecules, whereas it is not a critical component for the activation of the GLP-1 receptor by native GLP-1. These mechanisms are similar in that both induce the transition in the conformation of GLP-1 receptor through van der Waals interactions and hydrogen bonding of Trp33ECD with extracellular loop (ECL) 1 and ECL2, instead of direct interaction of the peptide with ECL2 [43]. Collectively, all GLP-1 receptor agonists, including native GLP-1, peptides, and small molecule GLP-1 receptor agonists, stabilize the binding pocket of the GLP-1 receptor in a similar fashion, which induces the TM helices 6 kink, a distinctive feature of the active state of the class B1 GPCR. Subtle differences with respect to interactions with the ECLs—specifically, the TM 6-ECL3-TM7/TM1—have led to biased agonism (i.e., selective agonism of the cAMP signaling pathway); this is in contrast to native GLP-1 which is also capable of simultaneously activating the beta-arrestin pathway [44]. Consequently, PF-06882961 is speculated to mirror native GLP-1 more closely by showing partial agonistic activity at recruiting beta-arrestin as well, but hitherto the augmentation of cAMP by the canonical pathway is sufficient to stimulate insulin secretion [45]. TT-OAD2 is another small molecule that also exhibits biased agonism in a unique binding mode [46]. Recent advances in the identification of crystallized structure, the spectrum of binding sites, and the mechanism of biased agonism are now facilitating the discovery of small molecule GLP-1 receptor agonists, all of which have a molecular weight of less than 1,000 g/mol.
Insulinotropic activity upon oral intake of small molecules has been reported in vivo with cynomolgus monkeys for LY3502970 and PF-06882961, and with humanized GLP-1 receptor mice for TT-OAD2. Particularly, when a high dose (5.4 mg/kg) of LY3502970 was given, the insulin secretion was comparable to that induced by exenatide, and an anorexigenic effect was achieved with just 0.05 mg/kg of LY3502970. The only literature-reported phase 1 clinical study to date was recently conducted with PF-06882961. In this study, the observed half-life of PF-06882961 was 4.3 to 6.1 hours. None of the healthy adult participants experienced dose-limiting toxicity at doses ranging from of 3 to 300 mg. Nausea, vomiting, and decreased appetite were the most common adverse effects, but they were not associated with premature withdrawal from the study. Of note, the pharmacokinetic profiles of PF-06882961 in the fasting and fed states were comparable, potentially enabling a more amenable dosing regimen.
Further studies are warranted to assess the exact site of absorption and the optimal dosage for the desired action with tolerable adverse effects. Will small molecule oral GLP-1 receptor agonists eventually replace injectable GLP-1 receptor agonists? In this regard, any small molecule oral GLP-1 receptor agonist must show cardiovascular benefits, which is one of the most distinctive beneficial effects, particularly for long-acting GLP-1 receptor agonists [35,47–49]. Considering the short half-life of small molecule oral GLP-1 receptor agonists, perhaps approaches to increase the duration of small molecules with a modified-release formulation may lead to improvements in long-term cardiovascular benefits.
Go to :
CONCLUSIONS
With the established beneficial clinical profile of GLP-1 receptor agonist class, considerable efforts to develop GLP-1 receptor agonists in a noninjectable formulation are garnering attention. All GLP-1 receptor agonists with a different route of administration faithfully serve its role as an incretin hormone, stimulating insulin secretion on nutrient ingestion; however, its subsidiary but important effects of body weight reduction, CVD risk reduction, and pharmacokinetic profiles differ according to the structure-activity relationship (Fig. 1). A novel approach using orally absorbable peptidyl and non-peptidyl agonists is paving the way toward a new era of GLP-1 receptor agonist therapy.
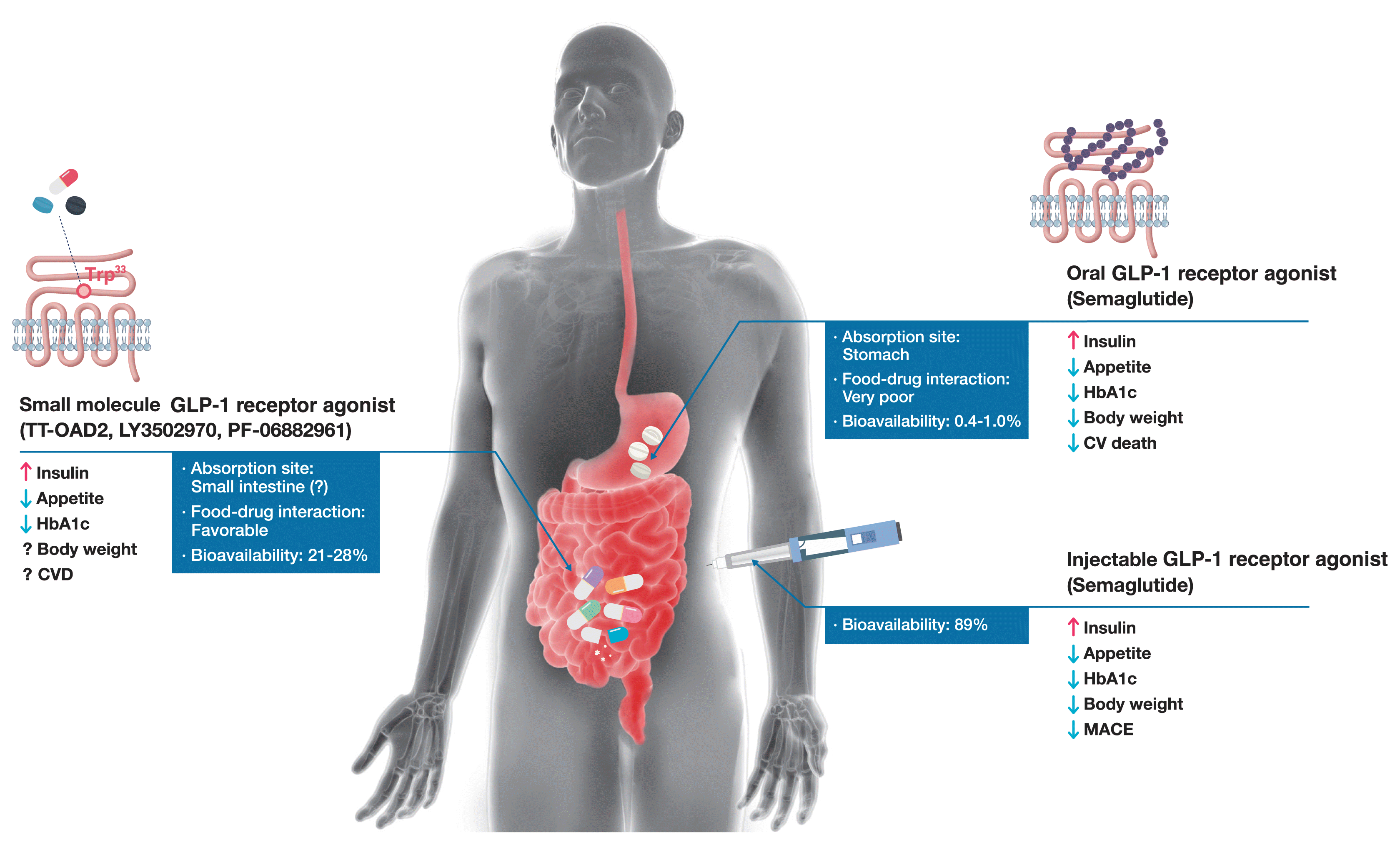 | Fig. 1Comparison of peptidyl and non-peptidyl glucagon-like peptide-1 (GLP-1) receptor agonists. The commercially available subcutaneous GLP-1 receptor agonists effectively elicit insulin response and control glucose metabolism with daily or weekly subcutaneous injections. Sustained GLP-1 signaling also possesses additional benefits: appetite and concomitant weight reduction, and cardioprotective benefits. The recently approved oral semaglutide should be taken with the recommended amount of water in the fasting state. Clinical trial results with oral and subcutaneous semaglutide revealed analogous properties, albeit different in magnitude. The left side of the figure describes small molecule GLP-1 receptor agonists currently in the process of drug development. These drugs are anticipated to overcome the challenges of oral semaglutide, most notably the substantial food-drug interactions and low oral bioavailability (0.4% to 1.0%). Dosing guidelines would be more liberal if these small molecules are absorbed in the small intestine with minimal food-drug interactions. As an example, LY3502970 showed a high bioavailability of 21% to 28% in cynomolgus monkeys. Long-term clinical outcomes regarding body weight reduction and cardiovascular outcomes are to be investigated. HbA1c, hemoglobin A1c; CVD, cardiovascular disease; CV, cardiovascular; MACE, major adverse cardiovascular event. aIn a phase 1 clinical study conducted with PF-06882961, the half-life and the area under the plasma drug concentration-time curve did not differ between the fed and fasted states. The authors infer that small molecule GLP-1 receptor agonists are absorbed mainly from the small intestine, as their physicochemical characteristics contrast with oral semaglutide, of which the absorption site is the stomach; bOral bioavailability of LY3502970 in cynomolgus monkeys. The bioavailability of small molecule GLP-1 receptor agonists in humans remains to be elucidated.
|
Go to :
 XML Download
XML Download