

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Osteoporosis is a major health problem in postmenopausal women [1]. In postmenopausal women, normal bone turnover cycles are impaired by estrogen deficiency, resulting in reduced bone mineral density (BMD) and increased risk of fracture in sites such as the spine, hip, and wrist [1]. According to the Korea National Health and Nutrition Examination Survey (2008 to 2011), the prevalence of osteoporosis in women aged ≥50 years is 38.0% [2]. The lifetime risk of osteoporosis-related fractures in women aged ≥50 years was 59.5% according to a Korean national health insurance review during 2005 to 2008 [3].
Treatment strategies to increase bone strength are essential to prevent postmenopausal women with osteoporosis from suffering a first or subsequent fracture [4]. For high-risk women with severe postmenopausal osteoporosis or previous fractures, one of the treatment options is to use anabolic agents, such as teriparatide and abaloparatide, to enhance bone formation [5]. However, in Korea, teriparatide was the only option as an anabolic agent until recently, and novel therapeutic options for severe osteoporosis to enhance BMD increase and reduce the risk of fracture, such as biologic agents, are warranted.
Romosozumab is a humanized monoclonal antibody directed against sclerostin, a glycoprotein produced by osteocytes [6]. Sclerostin is an endogenous inhibitor of the Wingless-type (Wnt) signaling pathway. Inhibition of sclerostin by romosozumab activates the Wnt signaling pathway in the bone, which results in an increase in bone formation and decrease in bone resorption, thereby resulting in a robust increase in BMD and possibly bone strength [7,8]. The efficacy of 210 mg romosozumab given as a subcutaneous once monthly (QM) injection has been demonstrated in various clinical trials. For postmenopausal women with osteoporosis, the efficacy of 12-month treatment with romosozumab in preventing fractures was demonstrated in the phase 3 Fracture Study in Postmenopausal Women with Osteoporosis (FRAME) [9] and Active-Controlled Fracture Study in Postmenopausal Women with Osteoporosis at High Risk (ARCH) [10] studies. Moreover, results from the An Open-label Study to Evaluate the Effect of Treatment With Romosozumab or Teriparatide in Postmenopausal Women (STRUCTURE) study showed that romosozumab demonstrated superior BMD gains over teriparatide at the total hip and femoral neck sites, as well as an increase in lumbar spine BMD [11]. In men with osteoporosis (the phase 3 PlaceBo-ContRolled Study EvaluatIng The Efficacy AnD Safety Of Romosozumab In TreatinG MEn With Osteoporosis [BRIDGE] study), treatment with romosozumab for 12 months was associated with a significantly greater mean percentage change from baseline in lumbar spine and total hip BMD compared with placebo [12]. Based on the results of these studies and the Korean phase 3 study reported in the current manuscript, romosozumab (Evenity™ Inj. Pre-filled Syringe, Amgen Korea Limited, Seoul, Korea) was approved in Korea for the treatment of osteoporosis in men and postmenopausal women at high risk for fracture [13].
This phase 3 study was conducted as a part of the regulatory requirements for the approval of romosozumab in postmenopausal women with osteoporosis in Korea, with the aim to evaluate the safety and efficacy of 6-month treatment with romosozumab (210 mg subcutaneously QM) in this patient population (ClinicalTrials.gov identifier NCT02791516).
METHODS
Study design
This phase 3, multicenter, randomized, double-blind, placebo-controlled study was conducted across 10 centers in Korea from January 2017 to September 2018. The dose and route of administration of romosozumab (210 mg subcutaneously QM) were selected on the basis of significant improvements from baseline in BMD outcomes demonstrated in previous phase 2 [14] and phase 3 [9,10] studies of romosozumab in postmenopausal women with osteoporosis.
After screening, eligible patients (postmenopausal women with osteoporosis) were randomized (1:1) to receive romosozumab (210 mg subcutaneously QM) or matching placebo for 6 months. Patients were followed up monthly for 6 months of treatment (primary analysis) and for an additional 3 months to assess the immunogenicity (formation of anti-romosozumab antibodies) and adverse events (AEs) for the final analysis (Fig. 1).
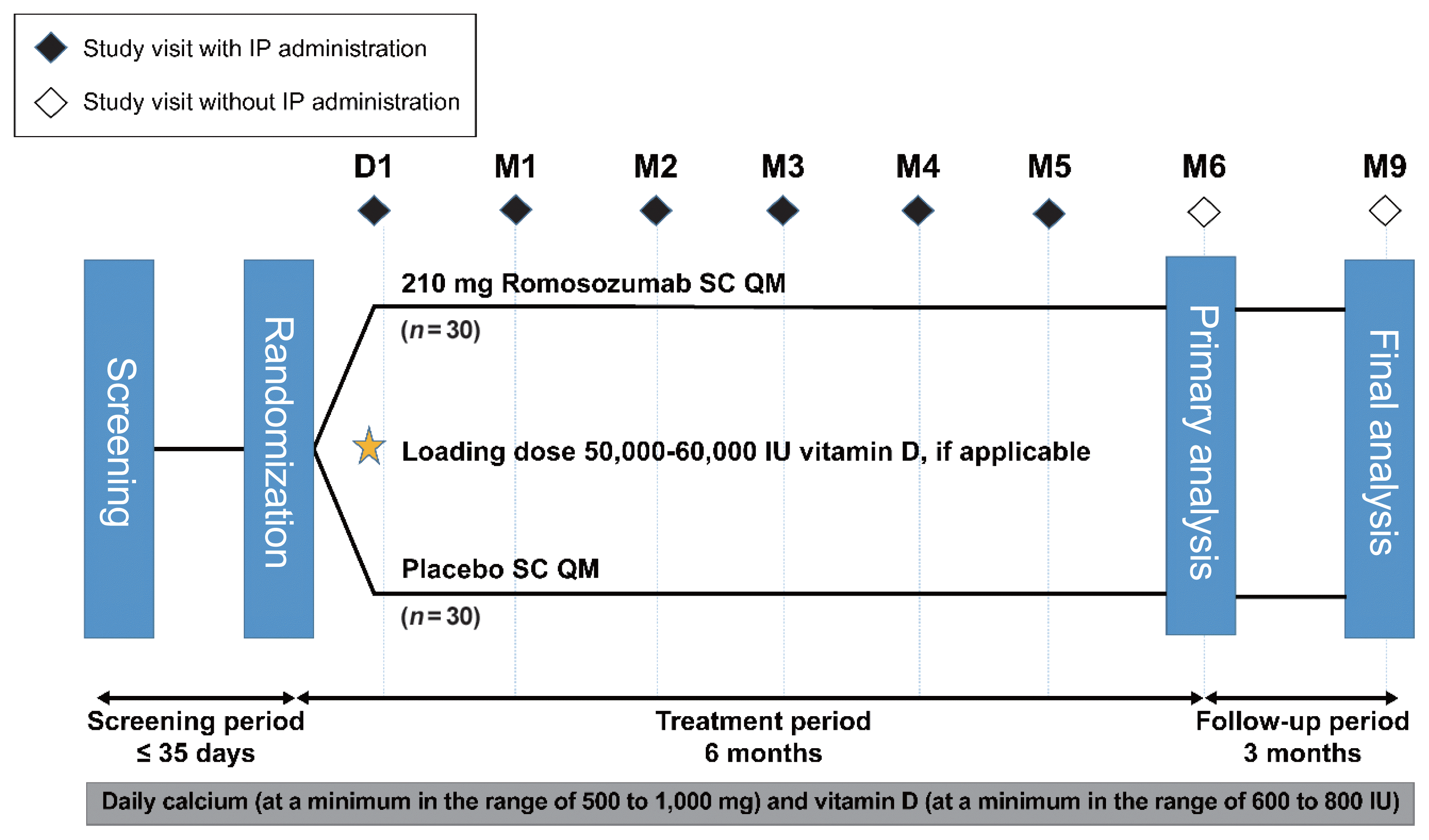
This study was conducted in accordance with the Ethical Principles for Medical Research Involving Human Subjects outlined in the Declaration of Helsinki in 1975 (revised in 2013), International Council for Harmonisation Good Clinical Practice (ICH GCP) guidelines, and other applicable regulations or guidelines. The study protocol and the informed consent form were approved by the Institutional Review Board at the Catholic University Yeouido Hospital (IRB no. 2016-1117-0035) and at each site. Prior to enrollment in the study, written informed consent was obtained from all patients, including consent for protocol-specific screening procedures and administration of study medication.
Participants
This study enrolled ambulatory, postmenopausal Korean women with osteoporosis (age ≥55 to ≤90 years). Patients with a BMD T-score ≤−2.5 at the lumbar spine, total hip, or femoral neck at the time of screening, as assessed by dual-energy X-ray absorptiometry (DXA), and at least two evaluable vertebrae in the L1 through L4 region and at least one hip evaluable by DXA were eligible for study participation. Key exclusion criteria were a BMD T-score ≤−4.0 at the lumbar spine, total hip, or femoral neck; history of hip fracture or metabolic or bone disease (except osteoporosis) that may interfere with the interpretation of the results; evidence of vitamin D insufficiency; history of solid organ or bone marrow transplant; current uncontrolled hyper- or hypothyroidism; current uncontrolled hyperparathyroidism or history of hypoparathyroidism; current hyper- or hypocalcemia; history of malignancy within 5 years (except nonmelanoma skin cancers and cervical or breast ductal carcinoma in situ); and possible diagnosis of multiple myeloma or related lymphoproliferative disorders. Patients who had previously taken osteoporosis medications were enrolled only if the washout period was sufficient (Supplemental Table S1).
Randomization and blinding
Eligible patients were randomized (1:1) to receive either romosozumab or placebo in a double-blind manner. Randomization was based on a schedule prepared by the sponsor’s central randomization group before the start of the study. Treatment randomization was performed via an interactive voice response system (IVRS). Information on the randomization was stored in the IVRS, and unblinded information was available only to the site staff authorized to access the IVRS. A patient’s treatment assignment was to be unblinded only when knowledge of the treatment was deemed essential for further management of the patient.
Interventions
During screening, patients underwent general, safety, laboratory (serum chemistry, hematology, serum 25-hydroxyvitamin (25(OH)) vitamin D levels, and serum protein electrophoresis), and imaging (lumbar spine and hip DXA scan) assessments.
Romosozumab (70 mg/mL, dosing volume 1 mL; Amgen Inc., Thousand Oaks, CA, USA) and matching placebo were supplied in single-use prefilled syringes and administered subcutaneously at the anterior abdominal wall, upper thigh, and upper arm using three prefilled syringes (cumulative dose of 210 mg romosozumab) on day 1 (on the day of randomization or within 72 hours of randomization) and months 1 to 5.
Patients with a serum 25(OH) vitamin D level of 20 to 40 ng/mL at screening received an initial loading dose of 50,000 to 60,000 IU vitamin D orally after randomization (within 1 week of the study day 1 visit). Those with a serum 25(OH) vitamin D level of >40 ng/mL at screening could also receive the vitamin D loading dose at the investigator’s discretion. Throughout the study period, patients received daily oral calcium (500 to 1,000 mg) and vitamin D (600 to 800 IU) supplementation.
The study treatment and assessments were discontinued when patients met any of the following criteria: patient’s request, AEs, ineligibility for study participation, protocol deviation, noncompliance to treatment, requirement for alternative therapy, protocol-specified criteria, death, lost to follow-up, and decision by the sponsor or investigator. In the event of a clinical fracture, the investigator was required to discuss the fracture risk and alternative treatment options, and patients were requested to complete the remaining visits regardless of the choice of post-discontinuation treatment. Study drug administration and other protocol-required therapies were also withheld or discontinued in patients with abnormal hepatic laboratory values, as specified in the Guidance for Industry Drug-Induced Liver Injury issued by the United States (US) Food and Drug Administration (FDA) [15].
Outcomes
The primary efficacy endpoint was the percent change from baseline in lumbar spine BMD at month 6. The secondary efficacy endpoints were the percent change from baseline in femoral neck and total hip BMD at month 6. Exploratory efficacy endpoints were the percent changes from baseline in bone turnover markers (BTMs; procollagen type 1 N-terminal propeptide [P1NP] as a bone formation marker and serum C-terminal telopeptide of type 1 collagen [CTX] as a bone resorption marker) over 6 months. Treatment-emergent adverse events (TEAEs), serious TEAEs, TEAEs of interest (hypersensitivity, cancer, hypocalcemia, injection site reaction, hyperostosis, osteoarthritis, atypical femoral fracture [AFF], osteonecrosis of the jaw [ONJ]), and serious cardiovascular events were monitored throughout the primary and final analysis periods as safety endpoints. Serum chemistry and hematology were assessed at screening, day 1, and months 1, 3, and 6 at Q2 Solutions (Singapore); blood anti-romosozumab antibodies were assessed at day 1 and months 1, 3, 6, and 9 at Amgen Inc. The pharmacokinetic endpoint was serum romosozumab concentrations assessed at day 1 and months 1, 3, and 6 at Amgen Inc.
Lumbar spine, femoral neck, and total hip BMD was assessed using DXA scan and centrally reviewed at Bioclinica (Portland, OR, USA). P1NP and CTX were assessed on day 1 and at months 1, 3, and 6. TEAEs were monitored throughout the study, and to fully evaluate the TEAEs of interest, potential events of ONJ, AFF, and serious cardiovascular TEAEs were submitted to an independent committee for adjudication.
Sample size
The sample size was determined based on the results from the phase 2 study of romosozumab in postmenopausal women [14]. This study found that the estimated mean percent difference in lumbar spine BMD between romosozumab and placebo at month 6 was 7.9% (95% confidence interval [CI], 6.6 to 9.3); the standard deviation (SD) of mean percent change in lumbar spine BMD at month 6 in the romosozumab and placebo groups was 3.9% and 3.8%, respectively (Amgen Inc., data on file). It was thus determined that a sample size of 30 patients per treatment group would provide >99% power to detect a significant treatment difference in the percent change in lumbar spine BMD at month 6, assuming a mean percent difference of 6.6% and an SD of 3.9 with two-sided type 1 error of 5% and a 10% dropout rate under the two-sample t test.
Statistical methods
All patients who were randomized, regardless of the treatment received, were included in the full analysis set. Among them, all patients who had a baseline DXA BMD measurement and at least one postbaseline DXA BMD measurement for the lumbar spine, femoral neck, or total hip skeletal sites were included in the BMD efficacy analysis set. The BTM efficacy analysis set included all patients who had a baseline measurement and at least one postbaseline measurement for CTX and P1NP. The safety analysis set consisted of all patients who received at least one dose of the study drug. All patients in the safety analysis set who had an evaluable serum romosozumab concentration were included in the pharmacokinetics analysis set.
Continuous variables were summarized descriptively using mean, median, SD, first quartile, third quartile, minimum, maximum, and the number of available observations. Nominal categorical variables were presented as frequencies and percentages. Percent change from baseline in lumbar spine, total hip, and femoral neck BMD at month 6 was evaluated using an analysis of covariance model. BTMs for treatment groups were presented as graphs depicting medians and interquartile ranges (IQRs) for percent change over time and assessed using the Wilcoxon rank-sum test. TEAEs were coded using Medical Dictionary for Regulatory Activities version 20.1 (primary analysis) or version 21.0 (final analysis). All statistical analyses were performed using SAS version 9.3 (SAS Institute, Cary, NC, USA).
RESULTS
Patient demographics and baseline characteristics
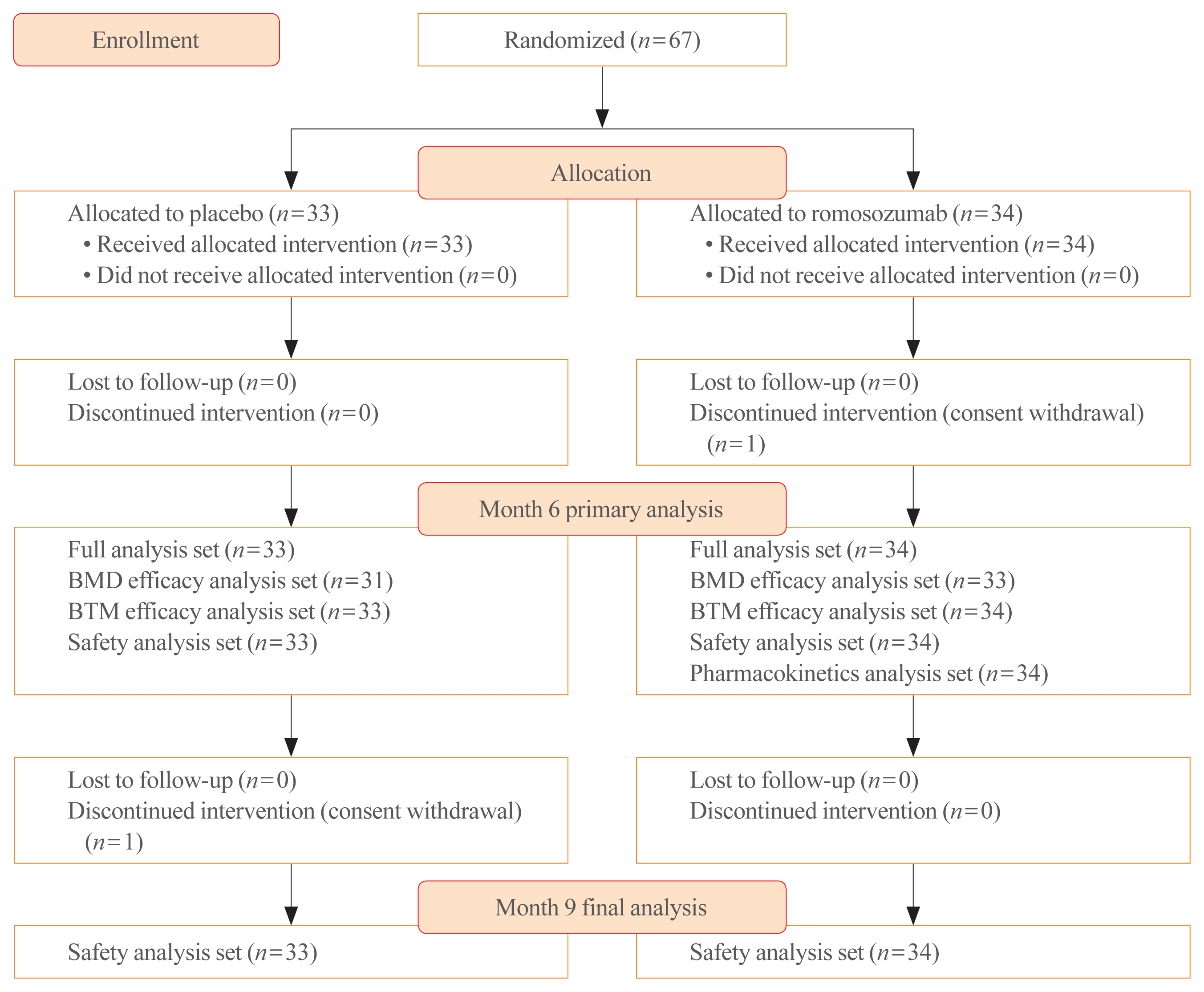
A total of 67 patients were enrolled and randomized into the romosozumab (n=34) and placebo groups (n=33). One patient in the romosozumab group and one patient in the placebo group withdrew their consent and discontinued the study before month 6 and 9, respectively. All the 34 and 33 patients in the romosozumab and placebo groups, respectively, were included in the full analysis set for the primary analysis at month 6 and the safety analysis set for the final analysis at month 9 (Fig. 2).
Patient demographic and baseline characteristics were generally balanced between the romosozumab and placebo groups, as illustrated by the age of patients (mean±SD, 66.7±7.6 and 68.4 ±7.2 years, respectively) and BMD T-score at the lumbar spine (−2.7±0.8 and −2.6±0.6, respectively) and total hip (−2.2± 0.7 and −2.2±0.6, respectively) (Table 1). Baseline laboratory parameters were also similar between the romosozumab and placebo groups (Supplemental Table S2). The distribution of baseline cardiovascular risk factors was similar between the groups. However, the proportion of patients aged ≥75 years was numerically higher in the placebo group than in the romosozumab group (seven patients [21.2%] vs. four patients [11.8%]), as was the proportion of patients with any history of cardiovascular risk factors (24 patients [72.7%] vs. 20 patients [58.8%]) (Supplemental Table S2).
Table 1
Patient Demographics and Baseline Characteristics (Full Analysis Set, Month 6 Primary Analysis)
| Variable | Placebo (n=33) | Romosozumab 210 mg SC QM (n=34) | All (n=67) |
|---|---|---|---|
| Age, yr | 68.4±7.2 | 66.7±7.6 | 67.5±7.4 |
|
|
|||
| Age group, yr | |||
| <65 | 13 (39.4) | 14 (41.2) | 27 (40.3) |
| ≥65–<75 | 13 (39.4) | 16 (47.1) | 29 (43.3) |
| ≥75 | 7 (21.2) | 4 (11.8) | 11 (16.4) |
|
|
|||
| BMI, kg/m2 | 22.8±2.3 | 23.4±2.9 | 23.1±2.6 |
|
|
|||
| Historical fracture | |||
| Any | 8 (24.2) | 7 (20.6) | 15 (22.4) |
| Osteoporotica | 6 (18.2) | 5 (14.7) | 11 (16.4) |
| Nonvertebral | 5 (15.2) | 5 (14.7) | 10 (14.9) |
| Major nonvertebralb | 5 (15.2) | 4 (11.8) | 9 (13.4) |
|
|
|||
| Any historical fracture at or after the age of 45 years | |||
| Any | 8 (24.2) | 7 (20.6) | 15 (22.4) |
| Osteoporotica | 6 (18.2) | 5 (14.7) | 11 (16.4) |
| Nonvertebral | 5 (15.2) | 5 (14.7) | 10 (14.9) |
| Major nonvertebralb | 5 (15.2) | 4 (11.8) | 9 (13.4) |
|
|
|||
| Time since the most recent fracture, moc | |||
| <12 | 0 | 0 | 0 |
| ≥12 | 8 (24.2) | 7 (20.6) | 15 (22.4) |
|
|
|||
| BMD T-score | |||
| Lumbar spine | −2.6±0.6 | −2.7±0.8 | −2.7±0.7 |
| Total hip | −2.2±0.6 | −2.2±0.7 | −2.2±0.7 |
| Femoral neck | −2.5±0.5 | −2.4±0.6 | −2.5±0.6 |
|
|
|||
| Serum 25(OH) vitamin D, ng/mL | 29.5±5.7 | 31.3±8.9 | 30.4±7.5 |
|
|
|||
| Serum 25(OH) vitamin D level, ng/mL | |||
| <20 | 0 | 1 (2.9) | 1 (1.5) |
| ≥20 | 33 (100.0) | 33 (97.1) | 66 (98.5) |
Values are expressed as mean±standard deviation or number (%). Percentages are based on the number of patients randomized.
SC, subcutaneous; QM, once monthly; BMI, body mass index; BMD, bone mineral density; 25(OH), 25-hydroxyvitamin.
a An osteoporotic fracture is defined as a fracture reported on the fracture electronic case report form (eCRF) page, excluding “skull fracture,” “facial bones fracture,” “fingers fracture,” “toes fracture,” fractures with high trauma severity, and pathological fractures;
![]()
All patients received concomitant calcium and vitamin D supplementation at baseline. Based on screening 25(OH) vitamin D values, an initial loading dose of 50,000 to 60,000 IU vitamin D was administered within 1 week of randomization to 32 and 33 patients in the romosozumab and placebo groups, respectively.
Efficacy
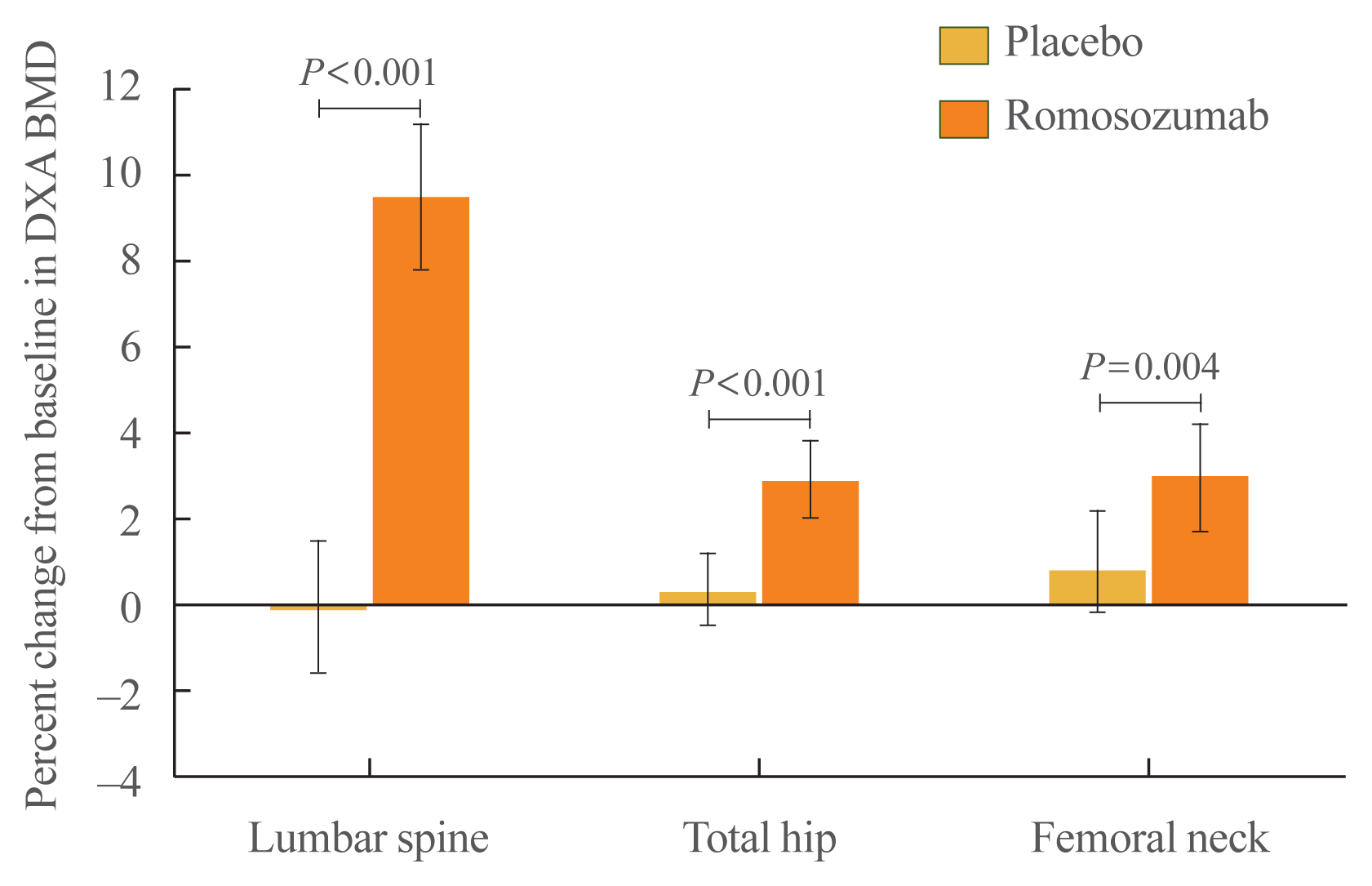
At month 6, the primary efficacy endpoint was met as the observed difference in the least square (LS) mean percent change from baseline in lumbar spine BMD between the romosozumab (9.5%; 95% CI, 7.8 to 11.2; n=33) and the placebo (−0.1%; 95% CI, −1.6 to 1.5; n=31) groups was significant (9.6%; 95% CI, 7.6 to 11.5; P<0.001). Furthermore, the difference in the LS mean percent change from baseline in total hip BMD between the groups at month 6 was significant (romosozumab group [2.9%; 95% CI, 2.0 to 3.8], placebo group [0.3%; 95% CI, −0.5 to 1.2], difference [2.6%; 95% CI, 1.4 to 3.7]; P<0.001). The difference in the LS mean percent change from baseline in femoral neck BMD between the groups (romosozumab group [3.0%; 95% CI, 1.7 to 4.2], placebo group [0.8%; 95% CI, −0.7 to 2.2], difference [2.2%; 95% CI, 0.7 to 3.6]; P=0.004) was also significant (Fig. 3).
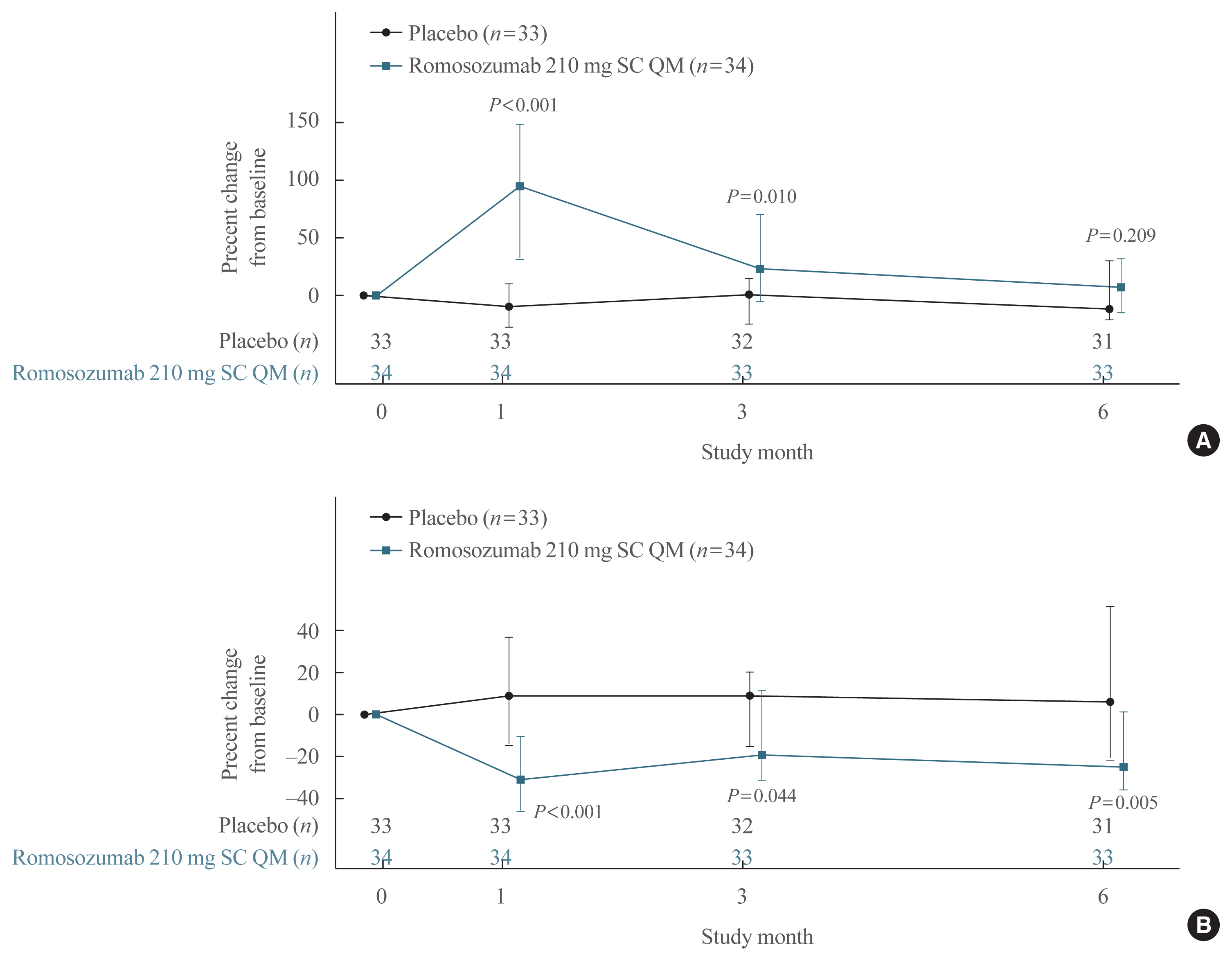
The percent change from baseline in P1NP in the romosozumab group showed a rapid significant increase compared with the placebo group at month 1 (median, 95.5% [IQR, 31.1 to 148.3] vs. −10.7% [IQR, −28.2 to 10.3], P<0.001), remained significantly high at month 3 (23.1% [IQR, −5.6 to 70.7] vs. 1.1% [IQR, −25.2 to 13.9], P=0.010), and returned to nearly baseline levels by month 6 (7.4% [IQR, −15.5 to 31.6] vs. −11.6% [IQR, −21.9 to 30.2]; P=0.209) (Fig. 4A). The median P1NP level at month 1 was 90.5 μg/L (IQR, 74.0 to 125.0) in the romosozumab group and 47.0 μg/L (IQR, 25.0 to 62.0) in the placebo group. The median percent change from baseline in serum CTX showed a sustained significant decrease in the romosozumab group compared with the placebo group at month 1 (−30.7% [IQR, −46.0 to −10.3] vs. 8.6% [IQR, −15.3 to 36.4], P<0.001), month 3 (−19.0% [IQR, −30.7 to 11.1] vs. 8.8% [IQR, −15.2 to 20.2], P=0.044), and month 6 (−25.2% [IQR, −35.8 to 1.3] vs. 6.1% [IQR, −21.6 to 51.2], P=0.005) (Fig. 4B).
Safety
During the 6-month treatment period, at least one TEAE was reported in nine patients (26.5%) in the romosozumab group and 13 patients (39.4%) in the placebo group. TEAEs reported in two or more patients were lumbar spinal stenosis (romosozumab, n=0; placebo, n=2) and nasopharyngitis (romosozumab, n=0; placebo, n=2) (Table 2). Serious TEAEs were reported in none of the romosozumab patients and in two placebo patients (lumbar spinal stenosis, grade 2; pulmonary tuberculosis and fracture femur, grade 3). No patients had TEAEs leading to treatment discontinuation or death.
Table 2
Summary of TEAEs (Safety Analysis Set, Month 6 Primary Analysis)
![]()
TEAEs of interest reported during the 6-month treatment period were hypersensitivity (rash in romosozumab, dermatitis contact in placebo, n=1 each), osteoarthritis (romosozumab, n=1), and hyperostosis (lumbar spinal stenosis, placebo, n=2). No events of cancer, hypocalcemia, injection site reaction, positively adjudicated AFF, positively adjudicated ONJ, or positively adjudicated serious cardiovascular events were observed. No femur fracture events were reported in the romosozumab group, and one patient in the placebo group reported a serious femur fracture (as noted above). No clinically relevant changes from baseline were observed for any of the hematology or chemistry laboratory parameters.
In the final analysis at month 9, the overall incidence of TEAEs was higher in the placebo group (48.5%) than that in the romosozumab group (35.3%). No patients had TEAEs leading to treatment discontinuation or death. No events of positively adjudicated AFF, positively adjudicated ONJ, or positively adjudicated serious cardiovascular events were reported during the final analysis period (Supplemental Table S3).
During the 6-month treatment period, five patients (14.7%) developed binding anti-romosozumab antibodies (cumulative: month 1 [n=1], month 3 [n=2], month 6 [n=5]), but no patient developed neutralizing antibodies. At the final analysis at month 9, six patients (17.6%) developed binding anti-romosozumab antibodies (two patients [5.9%] transient) and one patient (2.9%) developed neutralizing antibodies (nontransient). One patient was antibody negative at the primary analysis but tested positive for binding, non-neutralizing anti-romosozumab antibodies at the final analysis. Another patient who tested positive for binding, non-neutralizing anti-romosozumab antibodies at the primary analysis tested positive for neutralizing antibodies at the final analysis.
DISCUSSION
The results of this phase 3, multicenter, randomized, double-blind, placebo-controlled study showed that monthly treatment with romosozumab significantly increased lumbar spine BMD compared with placebo at 6 months of treatment (difference between the LS mean percent change from baseline 9.6%) in Korean postmenopausal women with osteoporosis. Significant increases in total hip and femoral neck BMD were also observed in the romosozumab group compared with the placebo group. P1NP showed a rapid significant increase at month 1 in the romosozumab group compared with the placebo group, and the median P1NP level observed at month 1 in the romosozumab group (90.5 μg/L) was higher than the normal reference intervals of P1NP in Korean women (18.7 to 83.2 μg/L) [16]. In contrast, serum CTX showed a sustained significant decrease in the romosozumab group compared with the placebo group throughout the 6-month study period.
The efficacy results of the current study were largely consistent with those obtained in previous phase 3 studies of romosozumab in postmenopausal women with osteoporosis [9–11]. The LS mean percent changes in BMD at the lumbar spine, total hip, and femoral neck in our study (9.5%, 2.9%, and 3.0%, respectively) were similar to those observed at month 6 in the romosozumab group of the FRAME (9.7%, 4.7%, and 2.3%, respectively) [9] and STRUCTURE (7.2%, 2.3%, and 2.1%, respectively) [11] studies. Our results also showed a rapid increase and return to baseline levels in P1NP and a sustained decrease from baseline in CTX after treatment with romosozumab. These results were also consistent with those observed in the previous phase 3 studies [9–11], suggesting a dual effect of romosozumab in increasing bone formation and decreasing bone resorption. The results of the current study should not be considered a justification for the 6-month use of romosozumab in Korea as it was tested in only a small sample of 67 patients. Moreover, physicians are advised to consider switching to antiresorptive therapies following treatment with romosozumab to avoid a decrease in BMD, per the Korean [13] and US FDA [17] prescribing information.
Inhibition of sclerostin is associated with potential safety concerns, such as cardiovascular risks [6]. Indeed, during the 12-month double-blind period of the ARCH study, positively adjudicated serious cardiovascular AEs, such as cardiac ischemic events and cerebrovascular events, were more frequently observed in the romosozumab group (2.5%) than in the alendronate group (1.9%) [10]. Given the ARCH study results, use of romosozumab is contraindicated in Korea [13] or should not be initiated in the US [17] in patients who have had a myocardial infarction or stroke within the preceding year. However, the incidence rates of positively adjudicated serious cardiovascular AEs were generally balanced between the romosozumab and placebo groups in the FRAME study [9] and in its subgroup analysis of Japanese participants [18]. Additionally, in our study, no positively adjudicated serious cardiovascular AEs were observed. Physicians have to make the risk-benefit evaluation of romosozumab treatment for each patient. Further real-world safety data will help physicians assess the risk-benefit balance of romosozumab treatment in their patients.
In our study, 17.6% and 2.9% of patients in the romosozumab group developed binding and neutralizing anti-romosozumab antibodies, respectively, at the month 9 final analysis. There results were largely consistent with those obtained in previous phase 3 studies in postmenopausal women with osteoporosis [9–11]. In previous studies, the presence of binding or neutralizing antibodies did not have a detectable impact on the efficacy or safety of romosozumab [9–11]. In our study, no notable effect on the drug exposure was observed in patients who tested positive for binding or neutralizing antibodies.
This study is the first to assess the efficacy and safety of romosozumab in postmenopausal women with osteoporosis in Korea. However, this study has some limitations. First, this study was conducted in a small population of 67 patients. Second, the rate of fracture risk reduction was not evaluated in this study. Third, the efficacy of romosozumab in increasing BMD was evaluated for only 6 months although romosozumab is recommended to be administered to patients for 1 year according to other pivotal studies [9–11]. Fourth, the effect of antiresorptive therapies following the 6-month romosozumab treatment could not be assessed in our study. Last, the current study results do not justify short-term (6-month) use of romosozumab in postmenopausal women with osteoporosis.
In conclusion, compared with placebo, treatment with romosozumab was well tolerated and significantly increased BMD at the lumbar spine, total hip, and femoral neck after 6 months of treatment in Korean postmenopausal women with osteoporosis. As with the previous pivotal studies, bone formation increased and bone resorption decreased immediately after treatment with romosozumab, and the bone formation returned to nearly baseline levels by month 6.
 XML Download
XML Download