

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
A histopathological classification of neoplasms reflects the biology and behavior of the tumors and serves as a guide for doctors and clinical management of patients. The World Health Organization (WHO) classification of tumors group publishes the WHO classification of tumors series to provide the international standards for cancer diagnosis including diagnostic criteria, pathological features, and molecular alterations. The WHO classification of tumors of endocrine organs was published in the fourth edition of the WHO series in 2017.
Compared with the third edition of the WHO classification published in 2004 [1], the most significant changes in the 2017 WHO classification of thyroid tumors involve (1) molecular and genetic characterization of follicular-derived thyroid tumors; (2) a new classification for encapsulated well-differentiated follicular tumors, in particular, introduction of tumors of borderline malignancy or uncertain malignant potential; (3) changing the international classification of diseases for oncology (ICD-O) behavior code of hyalinizing trabecular tumor to /1 (borderline or uncertain); (4) identification of new variants of papillary thyroid carcinoma (PTC); (5) subclassification of follicular thyroid carcinoma (FTC) into minimally invasive, encapsulated angioinvasive and widely invasive types; (6) reclassification of Hürthle cell adenoma/carcinoma and poorly differentiated thyroid carcinoma (PDTC) as distinct entities; and (7) changing the term carcinoma showing thymus-like differentiation to intrathyroid thymic carcinoma, as shown in Table 1 [2].
In the ICD-O codes, the morphology code records the histological type of the neoplasm and its biological activity in terms of how it behaves. Behavior codes /0, /1, /2, and /3 mean benign neoplasms, neoplasms of uncertain behavior, carcinoma in situ, and malignant neoplasm, respectively. The behavior code /2 has not been assigned to thyroid tumors. However, not all histologic types of the neoplasms have specific ICD-O code numbers. In the 10th revision of the International Statistical Classification of Diseases and Related Health Problems (ICD-10), the topography codes classify neoplasms according to their original site, assign malignant neoplasms to category C, and assign other neoplasms (in situ neoplasms, benign neoplasms, and neoplasms of uncertain or unknown behavior) to category D. Table 2 shows the ICD-10 and ICD-O codes of thyroid tumors according to the 2017 WHO classification.
In this review we briefly introduces a historical perspective on histopathological classification of thyroid tumors, changes in the fourth edition of the WHO classification of thyroid tumors, and update the contemporary diagnosis and classification of thyroid tumors since the revision of the WHO classification in 2017.
HOW THYROID TUMOR CLASSIFICATION SYSTEMS HAVE EVOLVED
There were revisions on diagnostic criteria for PTC in almost every edition of the WHO thyroid tumor classification. In the first edition (1974), PTC was a malignant epithelial tumor containing papillary structure (regardless of nuclear features). PTC-type nuclear features became essential criteria for malignancy in the second (1988) and third (2004) editions.
In the second edition, PTC was a malignant epithelial tumor showing evidence of follicular cell differentiation, typically with papillary and follicular structures as well as characteristic nuclear changes.
In the third edition, PTC was a malignant epithelial tumor showing evidence of follicular cell differentiation and a set of distinctive nuclear features. However, precise morphological criteria of distinctive nuclear features for PTCs were not provided to encourage a good consensus among pathologists, which resulted in severe interobserver variation in the diagnosis of PTCs.
In the fourth edition, a borderline tumor entity was incorporated to reduce the diagnostic discordance, and a nuclear score guide for RAS type PTC was provided. It was also aimed to reduce overdiagnosis and overtreatment of low-risk PTCs. The fourth edition further modified the definition of PTC and stated that “PTC is a malignant epithelial tumor showing evidence of follicular cell differentiation and a set of distinctive nuclear features. PTC is usually invasive. Papillae, invasion, or cytological features of PTC are required.” These changes were also intended to reduce overdiagnosis and overtreatment of low-risk PTCs, and the majority of encapsulated PTCs without clear-cut invasion were downgraded to the borderline tumor category, including well-differentiated tumor of uncertain malignant potential (WDT-UMP) and noninvasive follicular thyroid neoplasm with papillary-like nuclear features (NIFTP), because PTCs were still heterogeneous tumors genetically, and the nuclear feature alone was not successfully correlated with genetic alterations (RET/PTC rearrangements, BRAF V600E mutation, and RAS mutations) and accurate identification of the true biological nature of the tumors.
In the fourth edition WHO classification, FTC was classified into three prognostic groups: (1) minimally invasive (capsular invasive only) FTC; (2) encapsulated angioinvasive FTC; and (3) widely invasive FTC. Furthermore, in the follicular adenoma (FA)/FTC lineage, significant numbers of low-risk FTC (minimally invasive FTC) were downgraded to the borderline tumor category (follicular tumor of uncertain malignant potential [FT-UMP]) using stricter criteria of capsular and vascular invasions. It was also intended to reduce overtreatment for low-risk FTCs.
FOLLICULAR ADENOMA AND BORDERLINE TUMORS
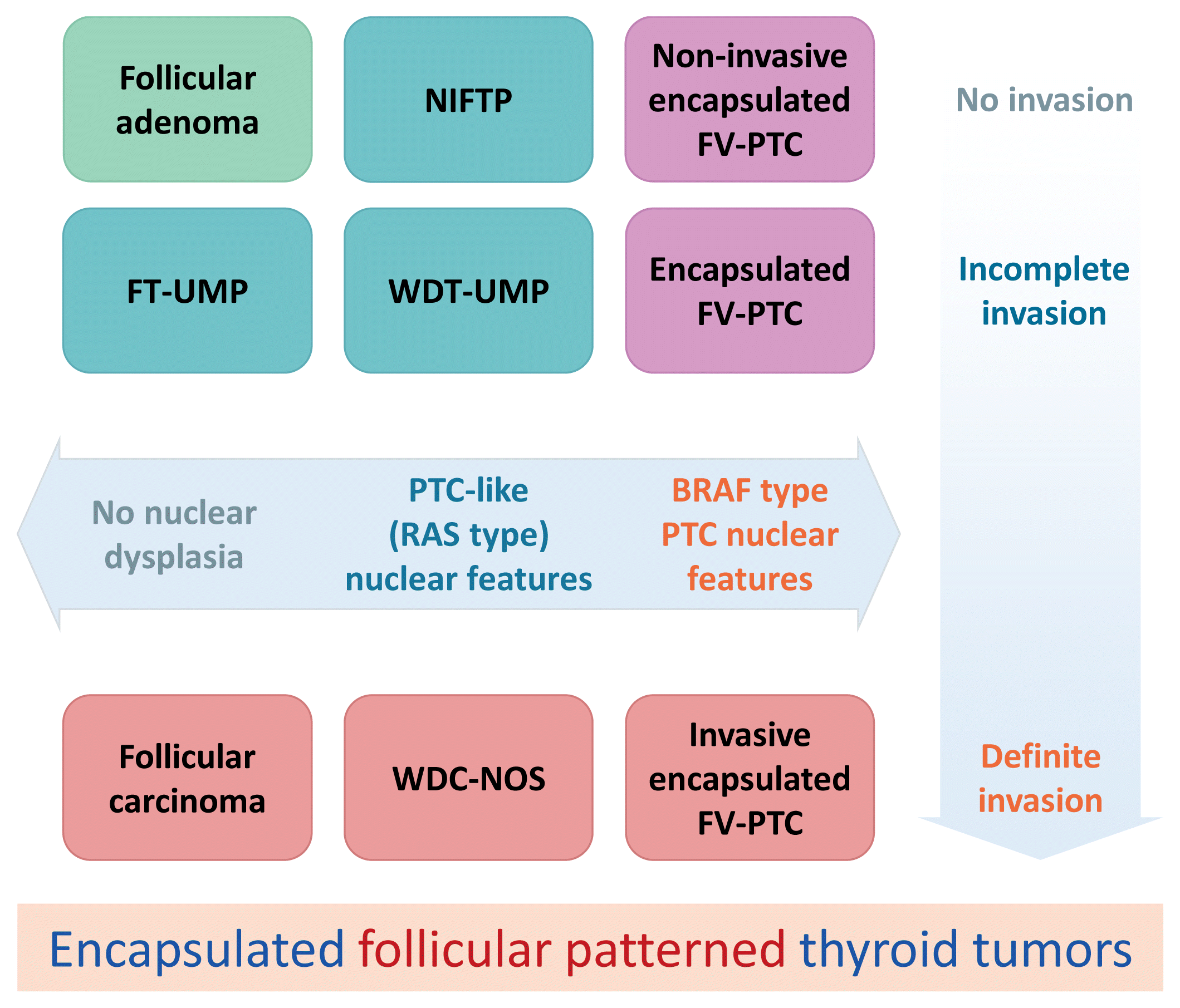
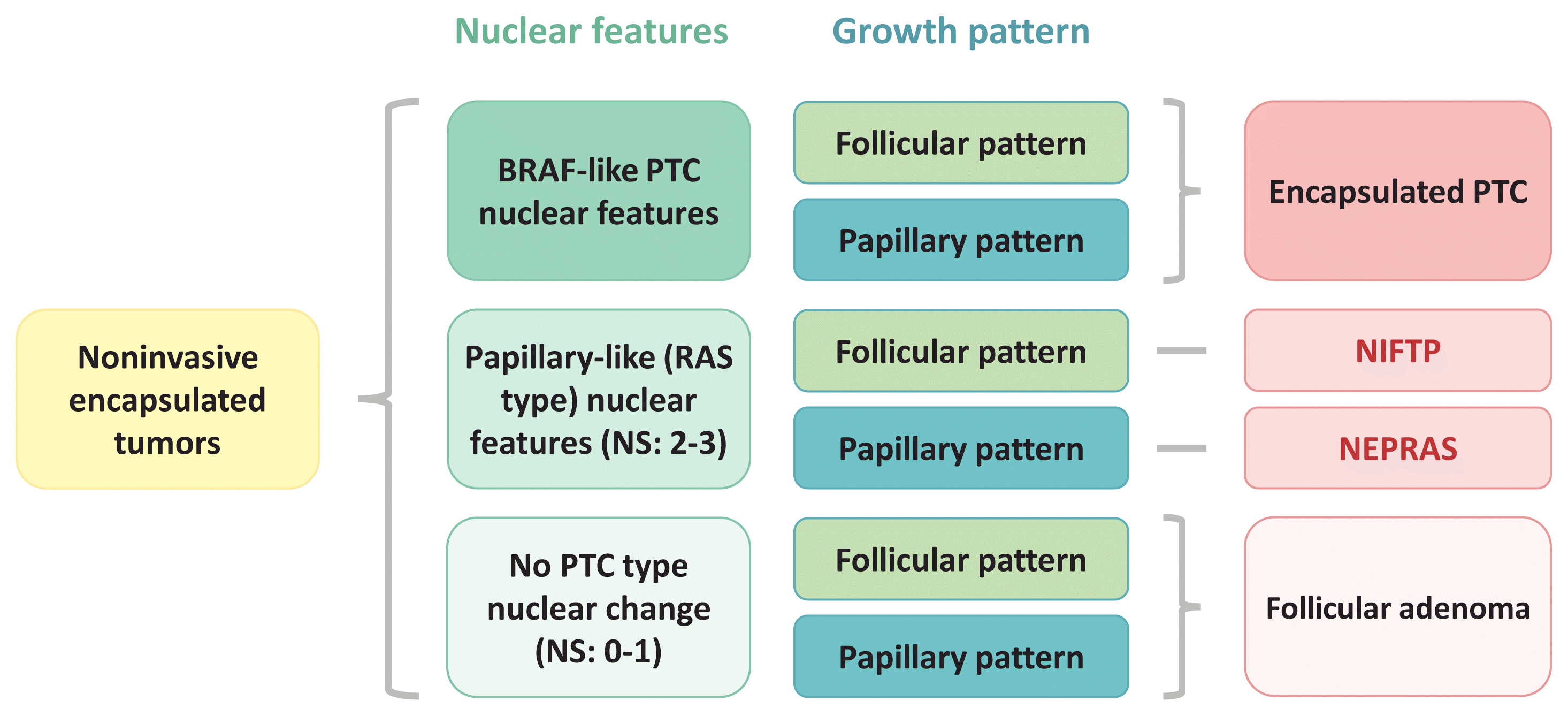
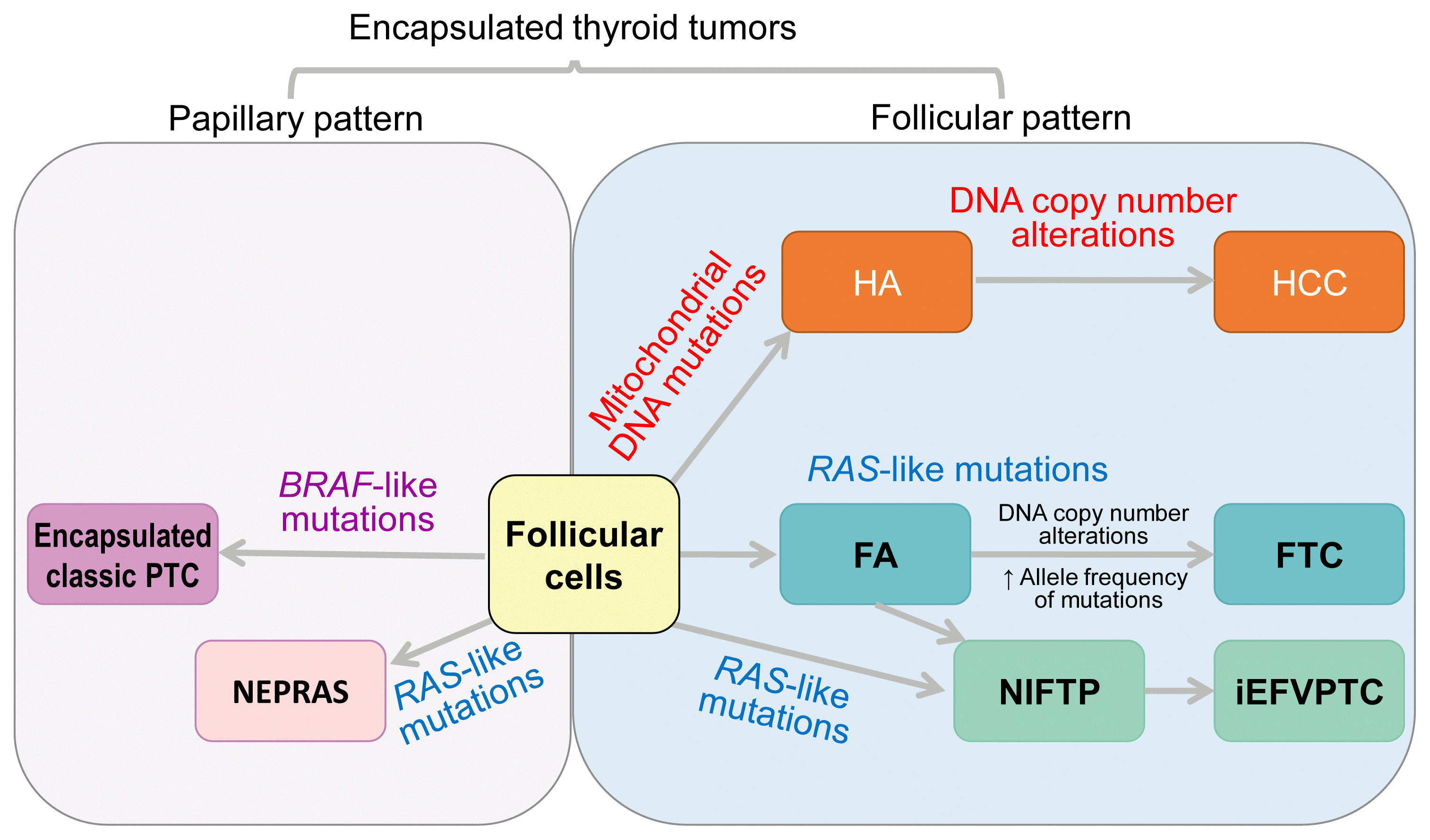
There were several breakthrough events in thyroid tumor classification recently. The introduction of borderline tumors (hyalinizing trabecular tumor, NIFTP, and tumors of uncertain malignant potential) to thyroid tumor classification is an epoch-making change in thyroid pathology practice [3–8]. With this breakthrough change, thyroid tumors are classified into three risk groups by the probability of recurrence/metastasis: negligible risk (<0.1%) in benign tumors, very low risk (<1%) in borderline tumors, and high risk in malignant tumors [2]. As a consequence, the definition of FA was altered in the new 2017 WHO classification of thyroid tumors [2]. It states that FA is a benign, encapsulated, noninvasive neoplasm showing evidence of thyroid follicular cell differentiation, without nuclear features of PTC [2]. A new phrase, “without nuclear features of PTC,” was added to the previous definition by the 2004 WHO classification of thyroid tumors, because noninvasive encapsulated follicular pattern tumors with PTC-like nuclear features are excluded from FA and are classified in NIFTP or WDT-UMP in the new definition (Fig. 1) [2].
There were several excellent articles and reviews that analyzed the effects of NIFTP on thyroid practice [2,7,9–22]. However, they focused on diagnosis, prevalence, and prognosis of NIFTP and did not emphasize the significant modification in the diagnosis of FA. Furthermore, several studies reported significant observer variation in PTC-type nuclear features and PTC-like nuclear features in the diagnosis of encapsulated follicular pattern thyroid tumors [21,23–25], which also should affect FA diagnosis.
In this review article, we would like to point out different handlings of the two types of nuclear features between Asian and Western thyroid practice [26]. They were called either (1) PTC-type nuclear features that have fully developed PTC-type nuclear changes (often observed in BRAF V600E mutated PTCs) and (2) PTC-like nuclear features (delicate nuclear changes) defined by Nikiforov et al. [8] using the nuclear score guide that were often seen in RAS mutated PTCs and NIFTP (Fig. 1). All of them were encapsulated follicular patterned thyroid tumors, and most review articles did not mention how to handle encapsulated papillary patterned tumors in detail, because tumors with true papillae were excluded from the NIFTP diagnosis in the definition [2,7,8,18]. However, differentiating between FA and noninvasive encapsulated PTC is a challenge for pathologists, because the differential diagnosis includes FA with papillary hyperplasia and noninvasive encapsulated classic PTC in addition to NIFTP, FT-UMP, WDT-UMP, and encapsulated follicular variant of PTC (Fig. 2).
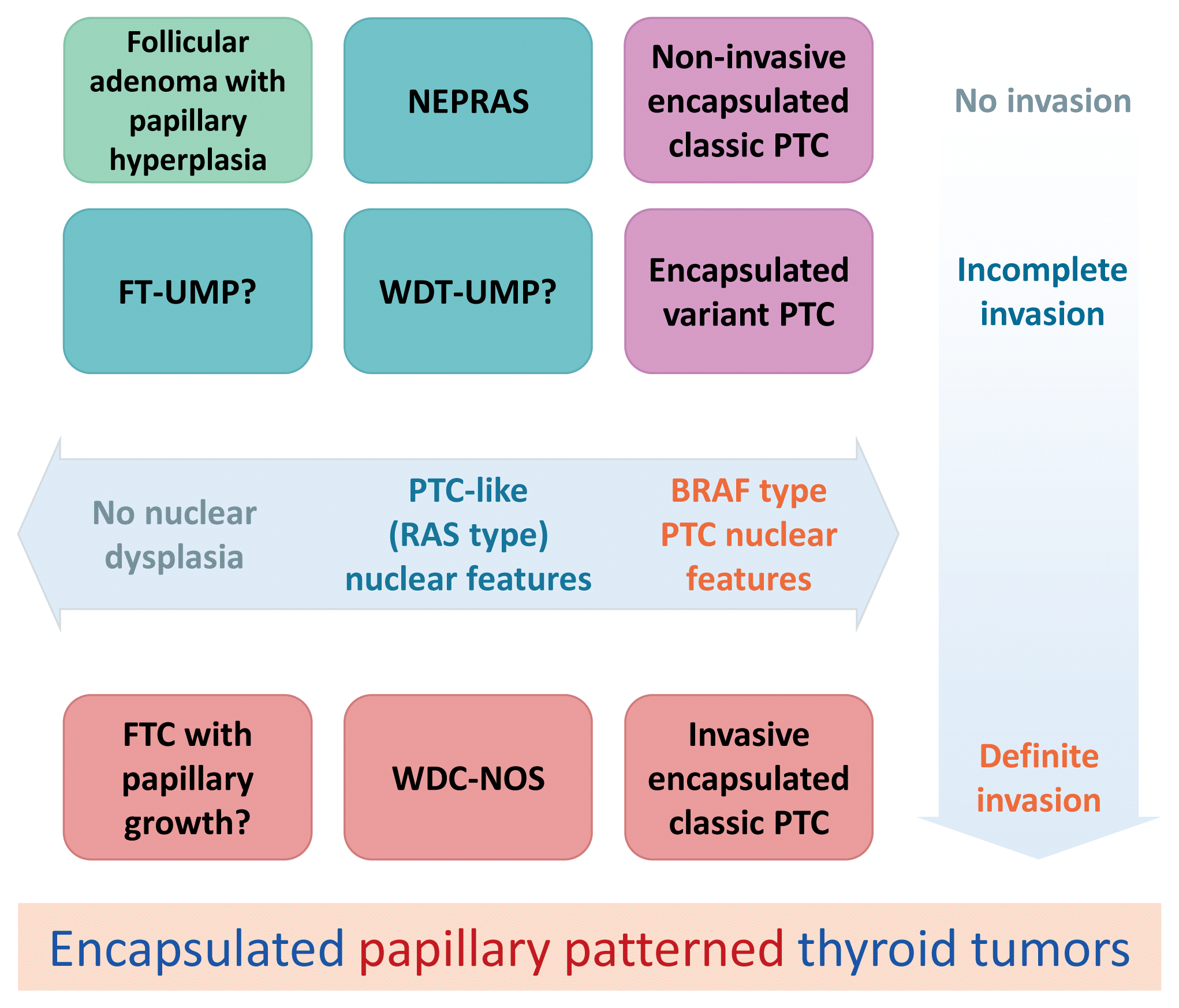
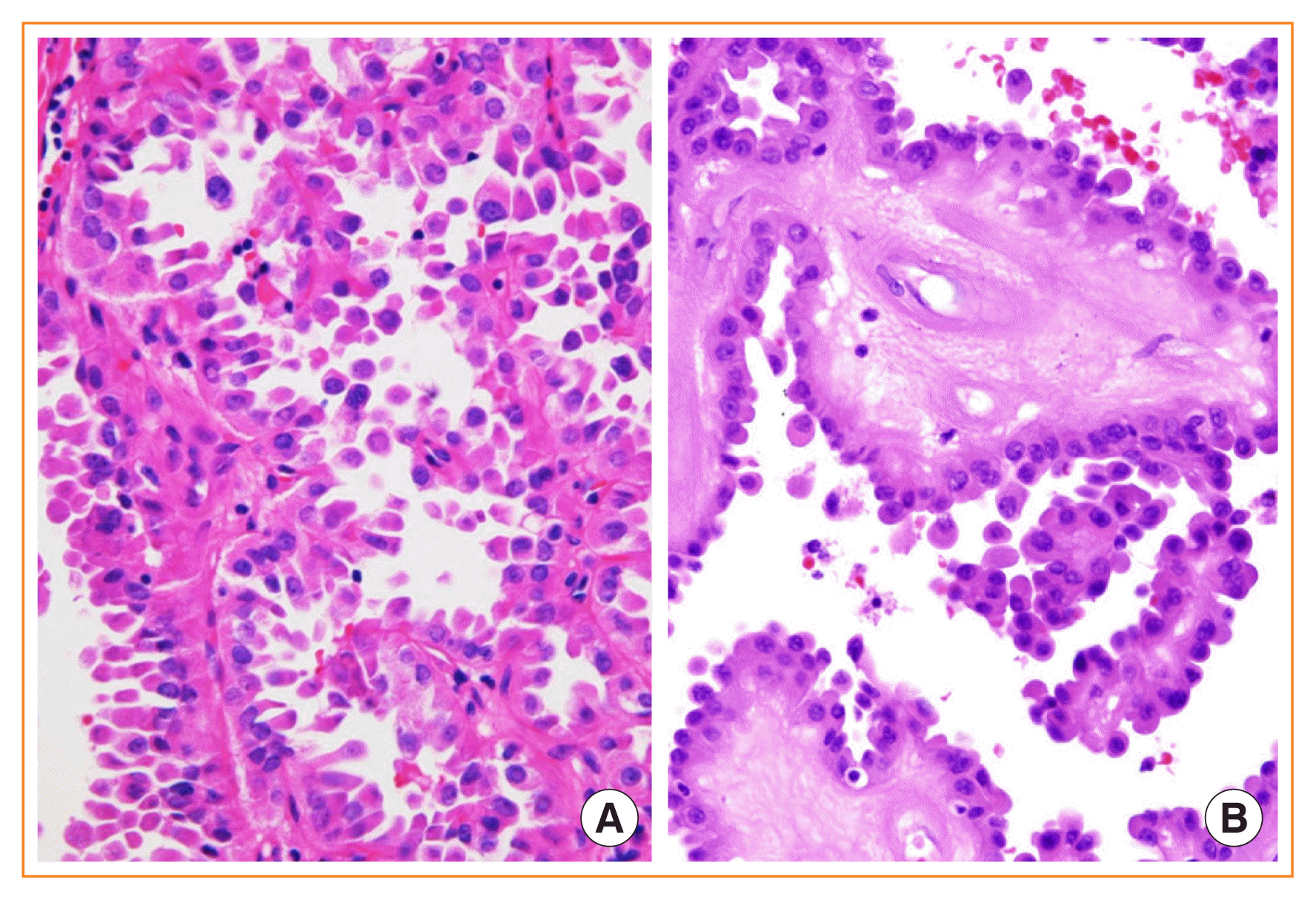
Observer variations in diagnosing those thyroid tumors have been reported to be significant [21,23–26]. There should be borderline/precursor thyroid tumors with a papillary growth pattern that was not clearly defined in the current WHO classification (Fig. 3) [27]. Ohba et al. [27] recently proposed a new borderline thyroid tumor with a papillary growth pattern and named it noninvasive encapsulated papillary RAS-like thyroid tumor (NEPRAS) (Fig. 4). Subsequent studies reported more cases of NEPRAS [28,29].
How to classify noninvasive encapsulated thyroid tumors into encapsulated PTC, NIFTP, NEPRAS, and FA, as we have recommended, is shown in Fig. 5 using nuclear features (BRAF-type PTC nuclear features or RAS-type papillary-like nuclear features) and growth patterns (follicular growth or papillary growth).
HYALINIZING TRABECULAR TUMOR
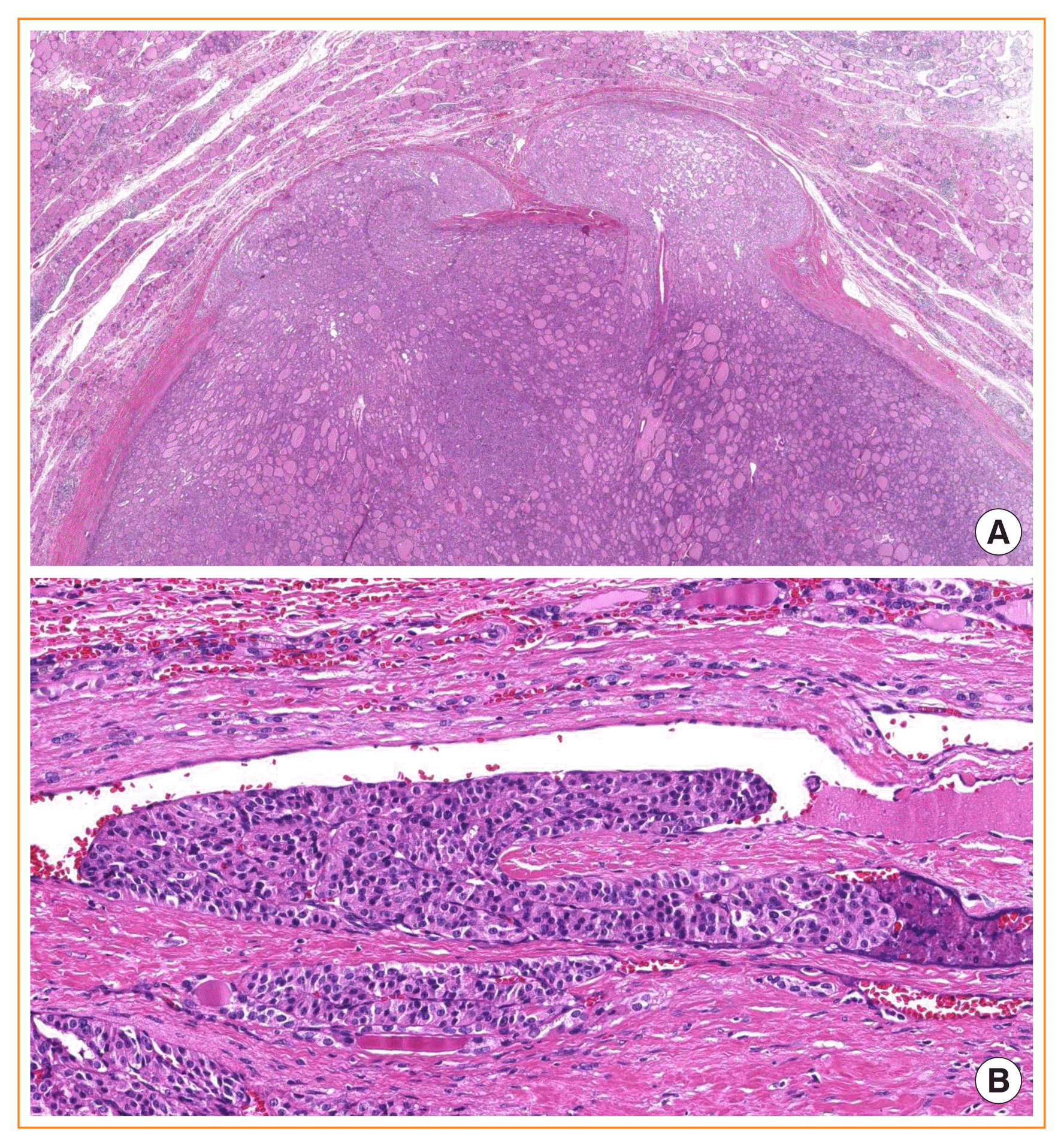
A hyalinizing trabecular tumor is a rare thyroid neoplasm of follicular cell origin with very low malignant potential. The tumor was moved from a benign group of “thyroid adenoma and related tumors” in the 2004 WHO classification to a separate category, where it is classified as a neoplasm of uncertain behavior with an ICD-O behavior code of /1 in the 2017 WHO classification [2]. Hyalinizing trabecular tumors have a very good prognosis. To date, only one case with lung metastasis has been reported [4], and regional lymph node metastases are reported in another case [30]. However, these cases with adverse outcomes have been criticized as a misdiagnosis of encapsulated solid/trabecular PTC.
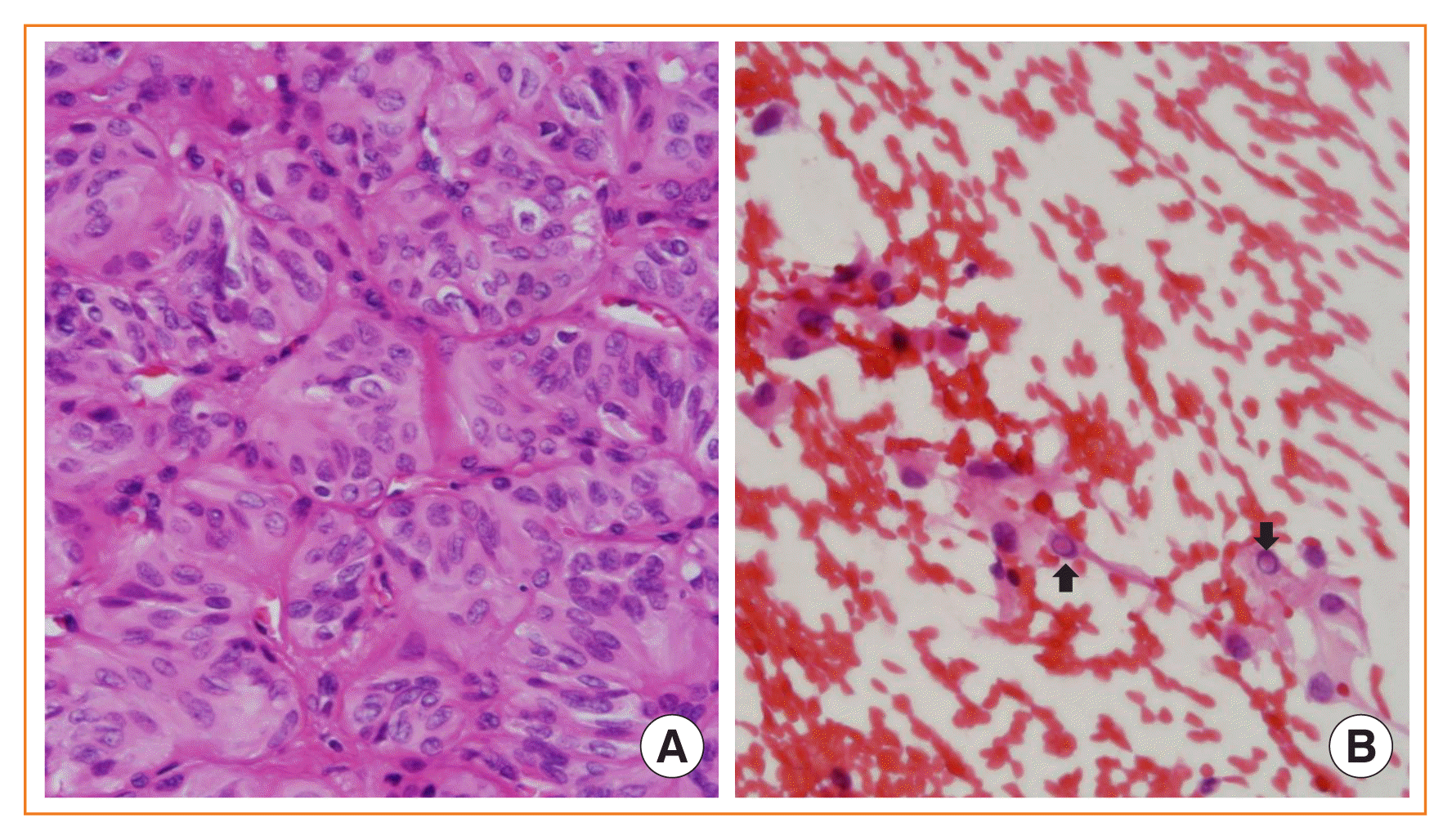
Because hyalinizing trabecular tumors have PTC-type nuclear features, including intranuclear grooves and pseudoinclusions, they can be challenging to diagnose in fine-needle aspiration cytology (Fig. 6). More than half of the tumors were diagnosed as PTC or suspicious for PTC [31].
Although RET/PTC fusions characteristic of PTC were reported in early studies [4], subsequent studies confirmed no RET/PTC rearrangements in the tumors using more accurate molecular diagnostic methods [31]. Recent studies reported the PAX8/GLIS fusions to be a genetic hallmark of hyalinizing trabecular tumor [31,32]. No PAX8/GLIS fusions have been found in other types of thyroid tumors to date [31–34]. Because all hyalinizing trabecular tumors with fully developed histopathologic features of the tumor had PAX8/GLIS fusions and benign clinical behavior, Nikiforova et al. [34] suggested that the diagnostic term for these tumors should be “GLIS-rearranged hyalinizing trabecular adenoma” changing the current term of “tumor” to “adenoma.”
PAPILLARY THYROID CARCINOMA
PTC had been the exclusive malignancy that could be diagnosed merely by its characteristics of cell nuclear features regardless of the tumor structures and growth patterns, until NIFTP was nominated and separated from the category of PTC, encapsulated follicular variant [2]. The addition of “PTC is usually invasive” in the definition of PTC suppresses the diagnosis of encapsulated tumors to PTC. This change may be in parallel with the construction of a new borderline tumor category. The diagnostic criteria for PTC allow it to demonstrate various histological features and growth patterns; so many PTC variants are recognized, including classic, microcarcinoma (≤1 cm in diameter), encapsulated, follicular, diffuse sclerosing, tall cell, columnar cell, cribriform-morular, hobnail, PTC with fibromatosis/fasciitis-like stroma, solid/trabecular, oncocytic, spindle cell, clear cell, and Warthin-like variants (Table 3) [2].
Among PTC variants, tall cell, columnar cells, and hobnail variants are of undoubted clinical significance, since they are aggressive variants associated with aggressive clinicopathological features and worse prognosis than for classic and encapsulated PTC [2,35,36]. In the 2015 American Thyroid Association risk stratification system, patients with these three aggressive variants are classified as having an intermediate risk of recurrence [37].
Although solid and diffuse sclerosing variants are regarded as aggressive variants of PTC by some researchers, their biological behaviors are still controversial [2,35,36]. An increase in the incidence of aggressive PTCs was observed at a rate higher than that seen in well-differentiated PTCs or anaplastic thyroid carcinomas (ATCs) in the past two decades in a study of a large cohort of thyroid cancers [36]. There is increasing evidence that when aggressive variants of PTCs present as low stage and without invasive features (including extrathyroid invasion, tumor thrombi, and lymph node metastasis), they might seem rather indolent [38,39].
Tall cell variant of PTC
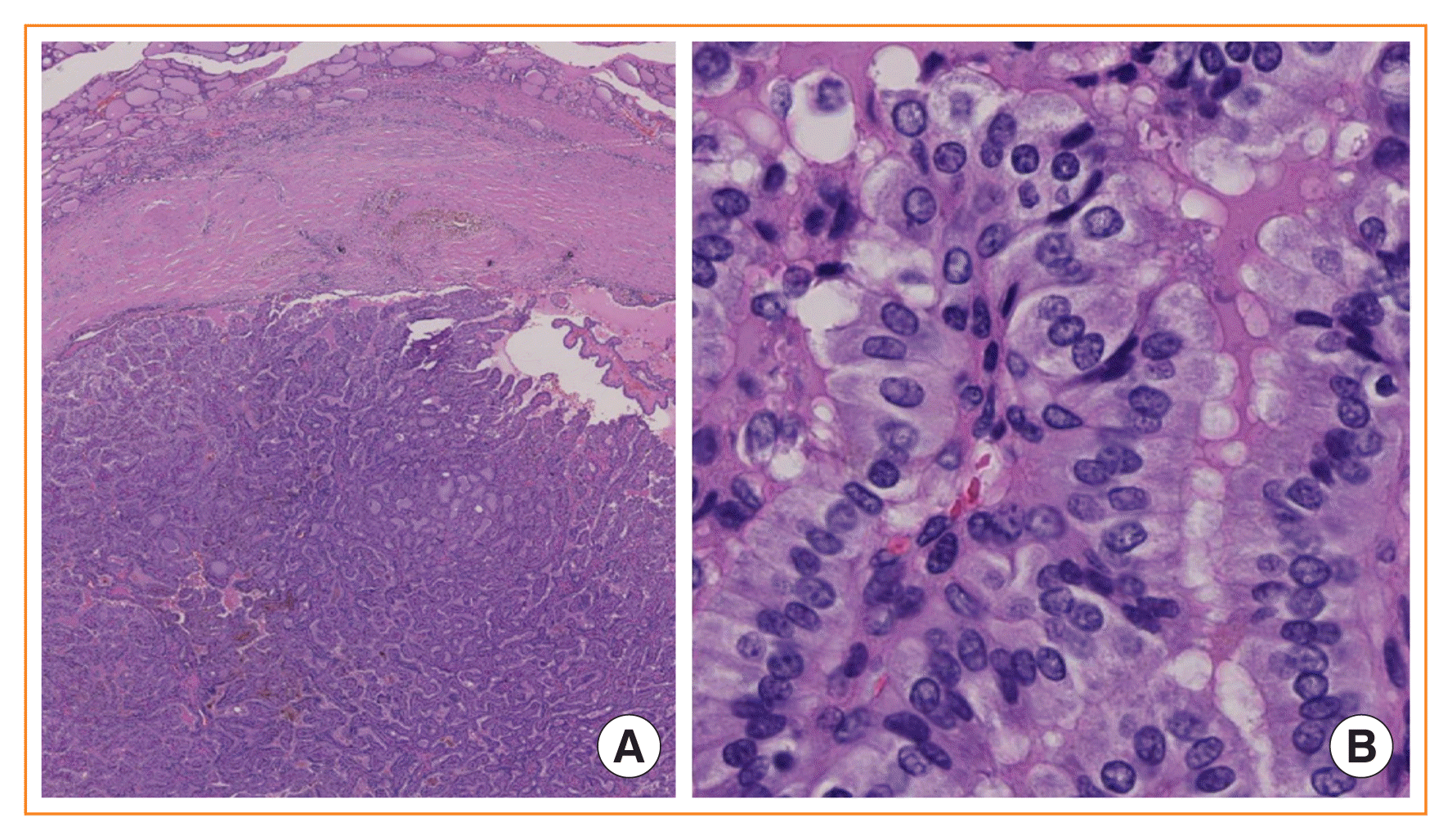
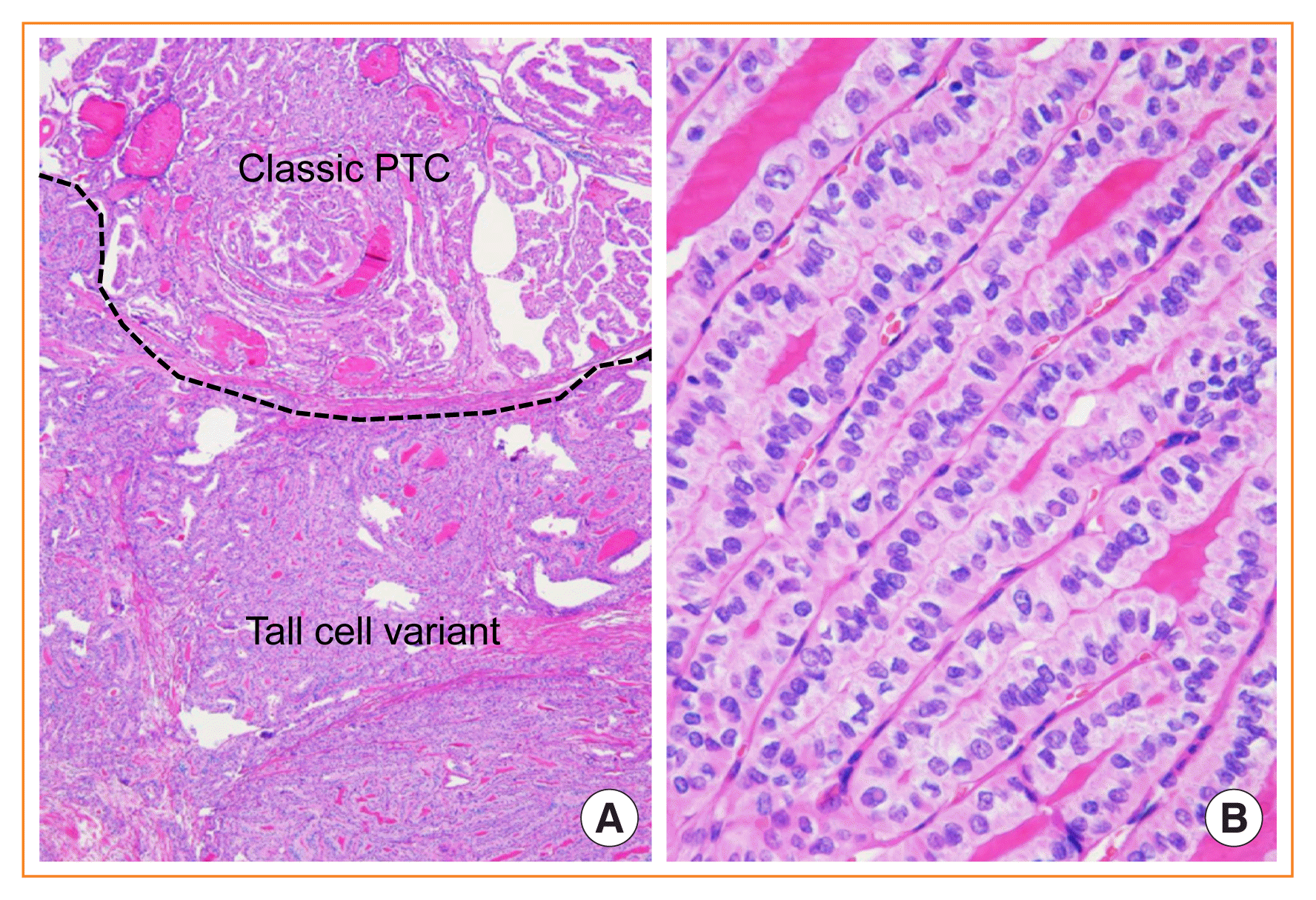
The definition of the tall cell variant was revised in the current WHO classification, because at least 30% of tumor cells are two to three times as tall as they are wide (Fig. 7), whereas in the former edition of the WHO classification, the tall cell variant needed to have predominant tumor cells three times as tall as they are wide [1,2,40]. The revision in the definition has allowed more cases of PTC to be included in this variant, and could predict disease-free survival more precisely [41,42].
There is increasing evidence that PTCs with as little as 10% tall cell change have a worse prognosis than do those without tall cells, and that PTCs with focal tall cell change (≥10%) should be reported and handled beyond the low-risk classification [41,43–47]. The diagnosis of the tall cell variant should not be missed, in that patients with this variant are more refractory to radioactive iodine ablation and have a worse prognosis than do those with classic PTC [36,48–50]. BRAF V600E and TERT promotor mutations are the most common genetic alterations for this variant [44,51].
Columnar cell variant of PTC
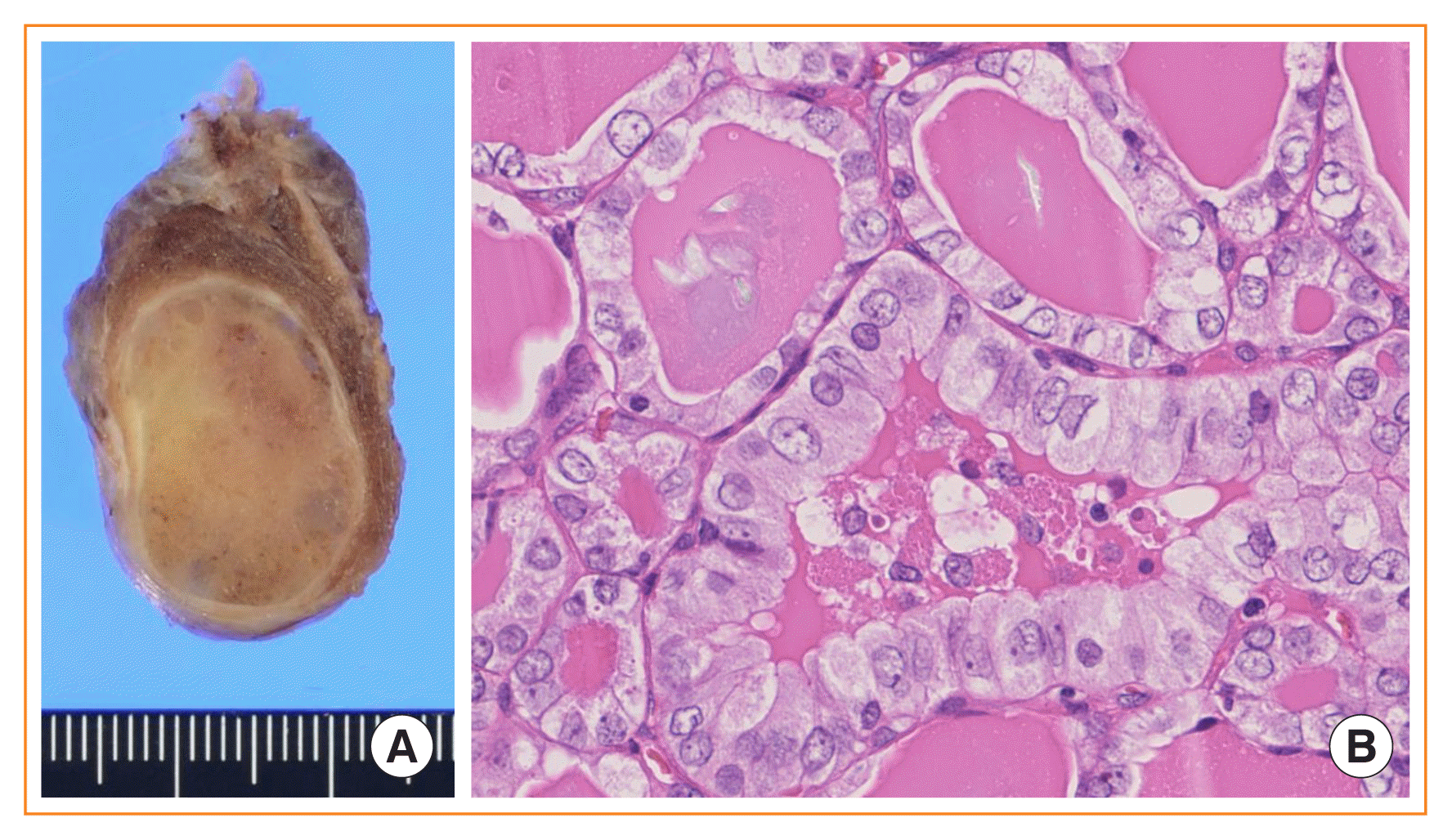
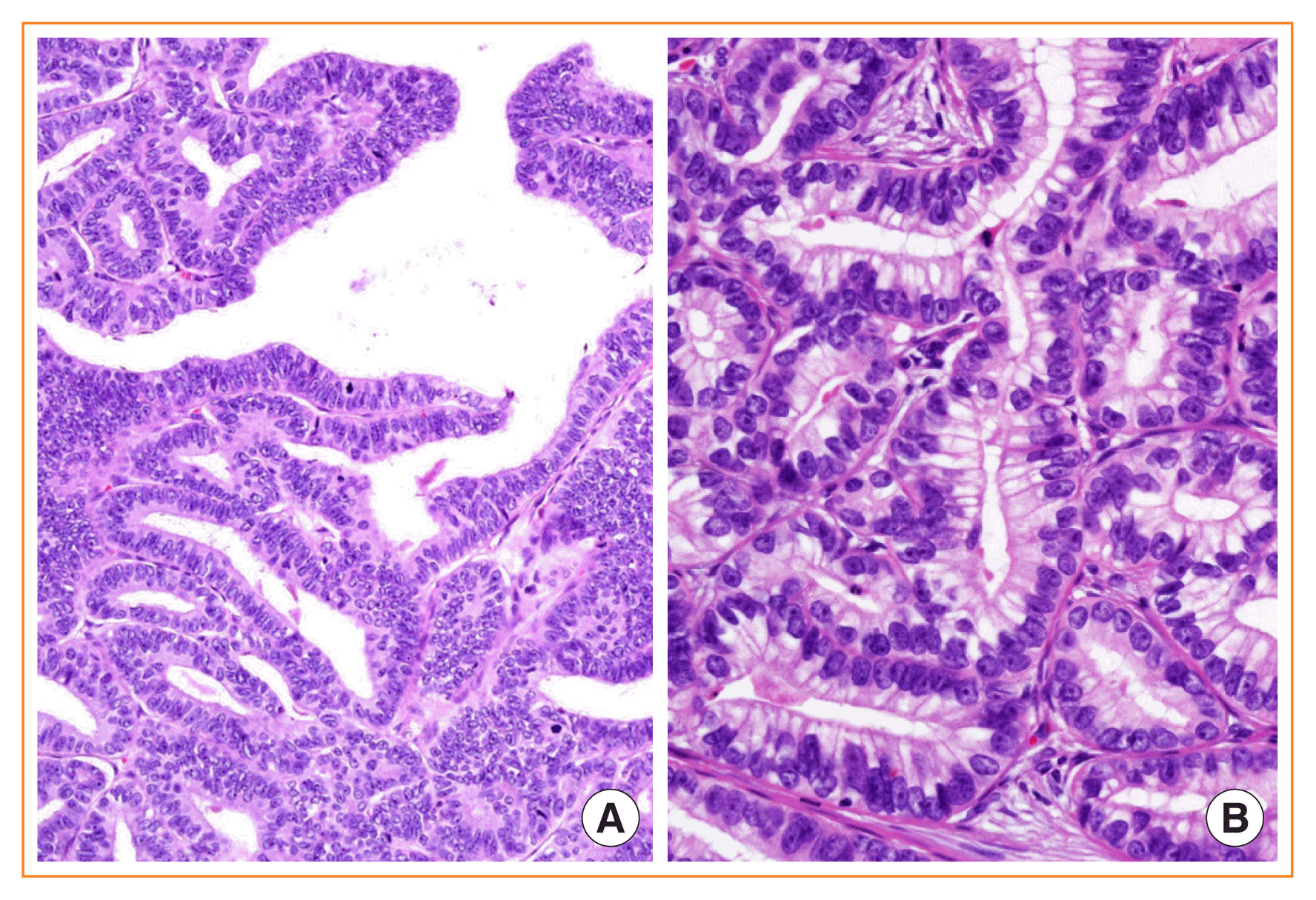
The columnar cell variant consists of columnar cells with marked pseudostratified hyperchromatic nuclei and absence of conventional PTC nuclear features (Fig. 8) [2,40]. It is an interesting finding that CDX2, a gut-specific nuclear transcription factor, the expression of which is a putative feature of intestinal-type differentiation, is positive in 10% to 55% of this variant [52,53], which may behave indolently or aggressively depending on tumor encapsulation and tumor size [54,55]. Columnar cell variants of circumscribed or intrathyroidal types have an excellent prognosis, whereas invasive tumors with extrathyroidal extension have a less favorable prognosis [50,56].
Hobnail variant of PTC
The hobnail variant is a novel entity in the 2017 WHO classification of tumors of endocrine organs, defined by >30% of tumor cells with hobnail features [2]. It is rare and accounts for <3% of all PTC cases [57]. Hobnail features in PTC were firstly described by Kakudo et al. [58] in 2004 as loss of cellular polarity and cohesiveness, with the main growth pattern of micropapillary and papillary structure, which was correlated with a higher risk of recurrence. In the following years, several articles concerning loss of cellular polarity and cohesiveness, micropapillary, or hobnail features in PTC were published, further clarifying that this feature represented an aggressive growth and was correlated with poor prognosis, in that most cases had lymph node/distant metastases and local recurrence, although different cut-off values of hobnail features were used in different studies, from two unequivocal points in the invasive front to 30% of total tumor cells [59–63]. An epithelial-mesenchymal transition was suggested as a possible mechanism of metastasis for this variant [60,62,64]. BRAF V600E mutation is the most common genetic alteration for this variant, accounting for 50% to 94% cases, followed by TP53 mutation [57,61,65–67].
The hobnail variant must be differentiated from classic PTCs showing hobnail-like morphology, which is associated with papillae with hyalinized and edematous fibrovascular core (Fig. 9) [68]. The hobnail-like morphology in classic PTC with cystic change is not a true hobnail variant but degenerative changes.
Solid variant of PTC
The solid variant should be identified when most of the tumor consists of solid/trabecular/nested components and other variants are ruled out [2]. A subset of solid variant PTCs, which lack conventional PTC nuclear features and present high mitotic figures and/or necrosis, fulfill the diagnostic criteria for PDTC and therefore are currently classified as PDTC [2,69].
This variant is strongly associated with ionizing radiation and is more common in young patients and children [70–73]. Radiation-associated pediatric cases frequently show RET/PTC3 rearrangement and sporadic cases show RET/PTC1/3 and ETV6/NTRK3 fusions [72,74]. BRAF V600E mutations are less commonly found than are classic PTC.
Diffuse sclerosing variant of PTC
The diffuse sclerosing variant, which usually presents with rapid and diffuse enlargement of the thyroid gland, clinically may be mistaken for autoimmune thyroiditis. It is most commonly seen in young women and is characterized by marked stromal fibrosis, dense lymphocytic infiltration, numerous psammoma bodies, and foci of squamous metaplasia, as well as frequent lymphovascular invasion [2,76]. RET/PTC rearrangements represented the genetic initiation event for this variant [77–79]. The BRAF V600E mutation is rare in this variant.
This variant is associated with extrathyroidal extension, cervical lymph node and distant metastasis, and shorter disease-free survival; however, the mortality rate is comparable to that of the classic variant [80–83]. The excellent long-term survival despite a higher recurrence rate may result from the favorable effect of a younger patient age.
FOLLICULAR THYROID CARCINOMA
FTC is the second common malignancy with follicular cell differentiation, which lacks the diagnostic nuclear features of PTC. In the 2004 WHO classification, FTC was divided into minimally invasive and widely invasive, according to the capsular invasion extent. In the 2017 WHO classification, minimally invasive FTC is further classified as minimally invasive (capsular invasion only) and encapsulated angioinvasive (with or without capsular invasion) (Fig. 10). There is no revision in the diagnostic criteria for widely invasive FTC, which extensively invades the thyroid gland and extra-thyroidal soft tissue and often shows extensive vascular invasion. However, extensive angioinvasion alone does not categorize the FTC as being widely invasive FTC.
The extent of vascular invasion in FTC is prognostically relevant [2,84–86]. In encapsulated angioinvasive FTC, tumors with limited vascular invasion (<4 vessels) have a better outcome than do those with extensive vascular invasion (≥4 vessels) [84,85]. However, extensive vascular invasion did not affect the survival of patients with widely invasive FTC independently [84]. The TERT promoter mutation was seen in FTC, but not in atypical FA or FT-UMP, and the TERT promoter mutation was significantly higher in patients with metastatic FTC than in patients with non-metastatic FTC [87].
HÜRTHLE (ONCOCYTIC) CELL TUMORS
Hürthle cell tumors, including Hürthle cell adenomas and Hürthle cell carcinomas, should be diagnosed if the majority of the tumor (>75%) consists of oncocytic cells [2,44]. Hürthle cell adenomas and carcinomas had been regarded as variants of FA and FTC, respectively, but they were nominated as a distinct entity in the 2017 WHO classification, since they have biological behaviors and molecular profiles different from those of their counterparts among follicular tumors (Fig. 11) [85,88–92]. Hürthle cell carcinomas are different from conventional FTCs, in that Hürthle cell carcinomas occur in older patients, are more common in men, can develop cervical lymph node metastasis, and have larger tumors, higher-stage disease, and worse prognosis, and are resistant to radioactive-iodine treatment [93]. As with conventional FTC, the prognosis of Hürthle cell carcinoma is related to the extent of vascular invasion [85].
Integrated genomic analysis of 56 Hürthle cell carcinomas revealed that, besides recurrent mitochondrial mutations, whole-chromosomal duplications of chromosomes 5 and 7 and widespread loss of heterozygosity because of haploidization and copy-number-neutral uniparental disomy were unique chromosomal landscapes for this special tumor [94]. EZH1 mutation was identified in a small subset of Hürthle cell adenoma and minimally invasive Hürthle cell carcinoma, but not in widely invasive Hürthle cell carcinoma [95].
POORLY DIFFERENTIATED THYROID CARCINOMA
PDTC is a follicular cell neoplasm that is morphologically and behaviorally intermediate between well-differentiated thyroid carcinoma (WDTC) and ATC. Different definitions of PDTC have been reported. The most well-known ones are Sakamoto-type PDTC, Memorial Sloan Kettering Cancer Center (MSKCC) criteria, and the Turin proposal [69,96,97]. Sakamoto et al. [96] focused on the tumor growth pattern (solid, trabecular, and/or scirrhous) in 1983. In the 2004 WHO classification, PDTC was vaguely defined as a “follicular-cell neoplasms that show limited evidence of structural follicular cell differentiation and both morphologically and behaviorally occupy an intermediate position between differentiated (follicular and papillary carcinomas) and undifferentiated (anaplastic) carcinomas” [1]. The MSKCC criteria defined PDTC based on the tumor necrosis and/or mitotic figures (≥5/10 high-power fields) in 2006 [97]. In 2007, the Turin proposal covered the definitions by Sakamoto et al. and MSKCC criteria, and made a revision [69,96,97].
The 2017 WHO classification adopted the Turin consensus proposal as the diagnostic criteria for PDTC, including (1) diagnosis of carcinoma of follicular cell origin; (2) a solid, trabecular, or insular growth pattern; (3) absence of conventional nuclear features of PTC; and (4) at least one of the following three features: convoluted nuclei, ≥3 mitoses per 10 high-power fields, or tumor necrosis [2,69]. The convoluted nuclei refer to raisin-like hyperchromatic nuclei with irregular membranes and may reflect dedifferentiation of PTC [69], but they are also considered by some pathologists to be degenerated nuclei without nuclear features of PTC.
Validation studies confirmed that the Turin consensus criteria could reliably select PDTC cases, and they reflected the patient prognosis more accurately than that in the 2004 WHO classification [13,98]. Although this algorithm was not initially used for Hürthle (oncocytic) cell carcinoma, it was proved to be practical and is now accepted as being useful for identifying oncocytic PDTC [2,99].
The Ki-67 index is a helpful marker used adjunctively in differentiating PDTC from WDTC and ATC, since it is usually 10% to 30% in PDTC, but <10% in WDTC and >30% in ATC [2,100–102]. Kakudo et al. [101] also proposed a prognostic classification of thyroid tumors into benign, borderline, and malignant tumors by the Ki-67 labelling index (Table 4). They proposed “high-risk thyroid carcinoma of follicular cell origin” defined by the Ki-67 index of 10% to 30% to cover all histological types of high-risk carcinomas, listed in the WHO classification as one of the synonyms and related terms of PDTC.
PDTC could be pure or could coexist with PTC or other types of tumors. In the latter case, the percentage of PDTC should be reported. As little as 10% of PDTC in a background of well-differentiated tumors could be associated with aggressive features and unfavorable prognosis [103]. The extent of invasion was an important parameter that affected clinical outcomes for patients with PDTC, and patients with encapsulated PDTC with capsular invasion only or focal vascular invasion (fewer than four foci) had an excellent outcome [104].
In accordance with its morphology and biological behavior, PDTC has genetic alterations embracing the alterations in PTC, FTC, and ATC: BRAF V600E, RAS, TERT promoter, TP53 mutations, and other high-risk gene mutations. The TERT promotor mutation represents the most common genetic alteration [105]. The prevalence of the TERT promoter mutation was significantly higher in tumors harboring aggressive histological features (including PDTC, tall cell variant of PTC, and widely invasive FTC) than in non-aggressive thyroid carcinomas, and TERT promoter mutations were associated with poor outcomes [106].
WELL-DIFFERENTIATED THYROID CARCINOMA WITH HIGH-GRADE FEATURES
Mitotic figures and necrosis are uncommon in PTC and FTC. Typically, mitoses are found to be less than three per 10 high-power fields in the WDTC. The presence of high mitotic rates and/or necrosis raises the possibility of PDTC. However, according to the WHO classification, the high-grade features alone do not categorize the tumor as PDTC (Fig. 12). The same tumors can be classified as PDTC by the MSKCC criteria if tumor necrosis and/or mitoses of ≥5 per 10 high-power fields are found in a follicular-derived carcinoma regardless of growth pattern [97,107].
ANAPLASTIC THYROID CARCINOMA
ATC is a highly aggressive malignancy that usually presents as advanced disease for which even radical surgery could not be possible. ATC can develop through dedifferentiation of a differentiated thyroid carcinoma or directly from follicular cells. Favorable patient survival was reported in cases with small foci of ATCs found incidentally within WDTCs after thyroid surgery [108]. Therefore, the percentage of ATC components should be noted in the pathology report.
Histologically, ATC can consist of one or in any combination of three patterns: sarcomatoid (composed of spindle cells), giant cell (composed of multinucleated giant cells), and epithelial (composed of squamoid or squamous tumor cells). All forms of ATC are characterized by a dense inflammatory infiltrate, tumor-infiltrating macrophages, necrosis, and a high mitotic rate. The rare histologic variants of ATC include paucicellular, rhabdoid, and small cell variants.
The diagnosis of ATC is challenging, since it overlaps morphologically with other malignancies showing anaplastic features, and the diagnoses should be made only from biopsy materials in most circumstances [102,109,110]. The rational use of thyroid-specific markers as well as other tissue-specific markers is helpful in differentiating ATC from its mimics [102,109,110]. PAX8 is reported to be a sensitive marker for detecting cells of thyroid origin other than TTF1 and thyroglobulin [109,111]. Recently, a multi-institutional study demonstrated that PAX8, detected by a monoclonal antibody MRQ-50 used worldwide, was positively expressed in 54.4% of 182 ATCs [112]. CD10 is diffusely and strongly expressed in ATC but absent or focally in WDTCs (Fig. 13) [113,114].
In a recent large retrospective cohort study of 360 cases of ATC, the TERT promoter mutation was most prevalent (75%), followed by TP53 (63%), BRAF (45%), RAS (24%), PIK3CA (18%), E1F1AX (14%), and PTEN mutations (14%) [115]. Concomitant TERT promoter and BRAF/RAS mutations were associated with worse outcomes than were mutation in only one of the genes [115].
THYROID TUMORS SHOWING THYMUS-LIKE DIFFERENTIATION
Thymic and third pharyngeal pouch remnants entrapped in the thyroid gland can give rise to ectopic thymoma, spindle epithelial tumor with thymus-like differentiation, and intrathyroid thymic carcinoma. The current 2017 WHO classification changed the term “carcinoma showing thymus-like differentiation” in the 2004 WHO classification to “intrathyroid thymic carcinoma” [1,2].
Intrathyroid thymic carcinomas share the immunohistochemical profile of primary thymoma that is positive for cytokeratin, CD5, KIT (also known as c-Kit and CD117), and p63, and negative for thyroglobulin and TTF1. Three histologic subtypes of this tumor include the squamous cell carcinoma type, lymphoepithelioma or basaloid type, and neuroendocrine carcinoma type.
NTRK-REARRANGED THYROID TUMORS
NTRK gene fusions found in the thyroid tumors involve NTRK1 and NTRK3 genes encoding the neurotrophin receptors TrkA and TrkC, respectively [33,116–118]. The most common NTRK fusion identified in PTC is ETV6-NTRK3 and less frequently identified fusions are TPM3-NTRK1, TPR-NTRK1, and TFG-NTRK1 [119]. The detection of ETV6-NTRK3 fusion is useful for the diagnosis of secretory carcinomas occurring in the breast, salivary gland, and thyroid gland. To date, all cases of secretory carcinomas of the thyroid gland have an ETV6-NTRK3 fusion only. Secretory carcinomas show various histologic patterns, including microcystic, cribriform, solid, papillary, and tubular growth. Colloid-like secretions are found in the intraluminal and cystic spaces. Tumor cells are positive for S100, mammaglobulin, GATA-3, and PAX8, but negative for thyroglobulin and TTF-1, helping to differentiate the tumor from PTC. Although this new entity has not been listed in the WHO classification of tumors of endocrine organs, cancer-type classification based on genetic alterations is clinically relevant, given that tyrosine kinase inhibitors targeting somatic mutations and fusions have been approved for the treatment of cancer patients [120].
CONCLUSIONS
Binary classification of benign or malignant thyroid neoplasms has raised many clinical problems of overdiagnosis and unnecessary treatment of thyroid cancers. The 2017 WHO classification categorized thyroid neoplasms into benign, borderline (uncertain malignant potential), and malignant tumors. Certain types of thyroid cancers were reclassified as borderline tumors. Histopathologic variants of PTC and FTC were redefined to better stratify prognosis and management of patients. Molecular-based classification for thyroid neoplasm allows diagnosis and prognostic risk stratification of thyroid cancer and may increase the accessibility of targeted treatments. As always, the current WHO classification of thyroid neoplasms will not be perfect, but it represents the current best-practice statements. Attempts to create a new classification system for thyroid tumors have also been made to improve the current risk stratification system for thyroid cancers.

 XML Download
XML Download