

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Chronic exposure to elevated lipid accumulation results in pancreatic β-cell apoptosis, induces hyperglycemia, and ultimately leads to the onset of type 2 diabetes [1]. Thus, protection of β-cells from damage caused by various external factors is an essential part of preventing the onset of diabetes. Although numerous studies have focused on various means for protecting pancreatic β-cells [2,3], a clear mechanism has not been identified thus far.
Autophagy is an intracellular self-degradation and rearrangement process to remove unnecessary or damaged organelles and maintain cellular homeostasis. It begins with the development of a double-layered lipid membrane, which besieges target organelles to form autophagosomes. In this step, microtubule-associated protein 1 light chain 3 (LC3) is conjugated with phosphatidylethanolamine (PE) and finally converted to LC3-II, which is known as an essential regulator of autophagy. Autophagosomes combine with lysosomes to form autolysosomes, and isolated target organelles are decomposed to amino acids by the lysosomal enzymes. Autophagy is generally considered to be a beneficial process for protecting cells against cell death and cellular stress, but it has also been recognized as a kind of cell death mechanism called “type 2 programmed cell death” [4]. The dysregulation of autophagy is implicated in various diseases, including neurodegenerative disorders, hepatic encephalopathy, and cancer [5]. The essential functions of autophagy in relation to diabetes have also been studied. Exposure to interleukin-1 induced severe damage in the late stage of autophagy as demonstrated by the accumulation of autophagic vacuoles in pancreatic β-cells [6], and defective autophagy was also detected in the β-cells of Zucker diabetic fatty rats as well as insulin-secreting cell lines following high glucose treatment [7]. In addition, impaired autophagy was observed in the β-cells of diabetic db/db and high-fat-fed C57BL/6 mice [8], and β-cell-specific knockout of the autophagy-related 7 (Atg7) gene, which regulates autophagy, resulted in β-cell dysfunction and apoptosis [9]. These studies suggest that autophagy is an essential mechanism for maintaining β-cell homeostasis and regulating the insulin secretion process.
Clusterin (CLU), also designated as apolipoprotein J (APOJ), testosterone-repressed prostate message2 (TRPM2), and sulfated glycoprotein 2 (SGP2), is a stress-activated chaperone-like heterodimeric glycoprotein that is ubiquitously expressed in various cells and affects cell adhesion, lipid transportation, membrane recycling, and tissue remodeling [10]. In addition, CLU has been reported to be associated with various diseases, such as cancer [11,12], Alzheimer’s disease [13], cardiovascular disease [14], and diabetes mellitus [15]. Upregulated expression of CLU has been detected in various cancers, and several studies have reported that CLU plays a role in cancer development and progression through cell survival and migration [16]. Recently, CLU has also been reported as a cytoprotective protein in disease-associated cells, including hepatocytes [17], neuronal cells [18], and cardiomyocytes [19]. However, the exact mechanism underlying how CLU exerts its cell protective effect is still unclear. Many studies have shown that CLU reduces mitochondrial apoptosis via interaction with BAX, activation of the Akt/NF-κB pathways, and repression of endoplasmic reticulum stress [17,20,21]. Thus far, although some potential mechanisms of the cytoprotective role of CLU have been reported, it appears to differ by cell type or stressors, and the exact molecular mechanism remains unclear.
In the present study, we investigated the role of CLU in autophagy regulation and its molecular mechanism in mouse insulinoma 6 (MIN6) cells. We also examined the effect of CLU on lipotoxicity-induced cell death and dysfunction.
METHODS
Cell culture and treatment
MIN6 cell lines (passages 18 to 20) were cultured at 37°C in a humidified incubator with 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 4.5 g/L glucose supplemented with 10% heat-inactivated fetal bovine serum, 100 IU/mL penicillin, 50 μg/mL streptomycin, and 50 μM 2-mercaptoethanol (all from Life Technologies, Paisley, UK). To examine the effect of CLU, the cells were incubated with CLU (Sigma-Aldrich, St. Louis, MO, USA), palmitic acid (R&D Systems Inc., Minneapolis, MN, USA), or a combination of both for 24 hours.
Small interfering RNA transfection
We designed small interfering RNA (siRNA) targeted against the mouse autophagy-related 3 (Atg3) gene using the siRNA target finder of GenScript. The sequences for mouse Atg3 siRNA were as follows: sense, 5′-CUG UCU UUG CUG UUA CGU U-3′ and antisense, 5′-AAC GUA ACA GCA AAG ACA G-3′ (Bioneer, Daejeon, Korea). The sense and antisense strands of the nonspecific siRNA duplex were as follows: sense, 5′-CCU ACG CCA CCA AUU UCG U-3′ and antisense, 5′-ACG AAA UUG GUG GCG UAG G-3′. Double-stranded siRNAs were transfected into MIN6 cells for 24 hours using Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific, Carlsbad, CA, USA) and then recovered in fresh media.
Immunoblotting analysis
Protein expression in cells was measured by immunoblotting analysis as previously described [22]. Briefly, total protein from MIN6 cells was prepared using radioimmunoprecipitation assay (RIPA) buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA) with protease and phosphatase inhibitors (Thermo Fisher Scientific). The proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. The following primary antibodies were used: anti-LC3, anti-β-actin, anti-Atg3, anti-Atg5, anti-Atg7, anti-SQSTM1/p62, anti-beclin1, anti-phosphor-mammalian target of rapamycin (mTOR), anti-Unc51 like autophagy activating kinase (ULK) 1, anti-cleaved caspase-3, and anti-total caspase-3 (Cell Signaling Technology, Danvers, MA, USA).
Immunofluorescence analysis
MIN6 cells were seeded in a glass bottom dish (SPL Life Science, Pocheon, Korea) and cultured for 24 hours in completed medium in the presence and absence of CLU and/or palmitic acid as described. The cells were fixed in ice-cold 4% paraformaldehyde for 5 minutes. Permeabilization was achieved in 0.01 M phosphate-buffered saline (PBS; Gibco, Bleiswjik, the Netherland) and 0.5% Triton X-100 (Sigma-Aldrich) for 5 minutes. After blocking with normal goat serum, the cells were incubated with the primary monoclonal antibody for LC3 (1:100; Cell Signaling Technology) for 24 hours at 4°C. The cells were then treated with a secondary fluorescein isothiocyanate (FITC) Alexa Fluor anti-rabbit antibody (1:400; Zymed, Carlsbad, CA, USA) for 1 hour at room temperature. To address the morphological relevance between LC3 and the other autophagy-related factors (Atg3 and p62), a double immunofluorescence assay was performed using MIN6 cells. To prevent mismatched binding between the antibodies for double labeling, two primary antibodies prepared from different animal species were applied simultaneously and detected by corresponding secondary antibodies conjugated with FITC or tetramethylrhodamine (TRITC). To visualize the nuclei, the MIN6 cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA, USA), and the detected proteins were observed and analyzed using an Olympus BX51 microscope (Olympus Optical Co., Tokyo, Japan).
Immunoprecipitation assay
Total protein samples from MIN6 cells were lysed using RIPA buffer and then collected for LC3 immunoprecipitation. The proteins were incubated with 2 μg of anti-LC3 antibody (Cell Signaling Technology) and stored at 4°C overnight. Bound proteins were recovered after the addition of 30 μL of protein A/G agarose beads (Invitrogen) for 1 hour at 4°C. The beads were gently centrifuged for 1 minute and washed three times for 5 minutes each. The bound proteins were resuspended in 20 μL of RIPA buffer solution. After heating at 95°C for 5 minutes, the immunoprecipitants were separated by SDS-PAGE and then transferred onto PVDF membranes for immunoblotting analysis to detect Atg3 and p62 proteins.
In situ detection of fragmented DNA (terminal deoxynucleotidyl transferase-mediated UTP nick end labeling assay)
DNA fragmentation was examined using a commercial kit for the detection of apoptosis (ApopTag Red, Merck, Temecula, CA, USA). After fixation and permeabilization, the cells were added to 75 μL equilibration buffer for 3 minutes, then exposed to 55 μL working strength terminal transferase enzyme solution in a humidified chamber at 37°C for 1 hour. After washing with stop/wash buffer, the cells were incubated with warmed anti-digoxigenin conjugate solution at 37°C without exposure to light. Finally, the cells were counter-stained with DAPI for 10 minutes and mounted on glass sliders after washing with PBS.
RNA extraction and real-time polymerase chain reaction analysis
Total RNA was extracted with TRIzol (Life Technologies, Carlsbad, CA, USA). The first strand of complementary DNA was synthesized by oligo(dT)-primed reverse transcription using an RT enzyme mix (Thermo Fisher Scientific, Rockford, IL, USA), and applied as a template for a real-time polymerase chain reaction (PCR) assay with SYBR green I Master (Roche Diagnostics, Indianapolis, IN, USA) and following primers: Atg3, sense 5′-TAAGGCTGACGCTGGAGGTGAA-3′ and antisense 5′-GTGCTCAACTGTTAAAGGCTGCC-3′, NM_ 026402). The PCR analysis was performed with a Light Cycler 480 (Roche, Lewis, UK) and the relative expression of RNA was measured by the ΔΔCt formula method.
Glucose-stimulated insulin secretion test
After pre-incubation with glucose-free Krebs Ringer bicarbonate HEPES (KRPH) buffer for 1 hour, MIN6 cells were exposed to DMEM containing 1 or 4.5 g/L glucose for 1 hour. The amount of released insulin was measured by a mouse insulin enzyme-linked immunosorbent assay (Linco Research, St. Charles, MO, USA).
Statistical analysis
The statistical data are expressed as the mean±standard error of the mean. All calculations were performed using SPSS Statistics version 12.0 software (SPSS Inc., Chicago, IL, USA). For evaluation of statistical differences, the Student’s t test or a one-way analysis of variance was used, and multiple comparisons among the experimental groups were investigated with the Bonferroni correction test. Differences with a P value <0.05 were considered significant.
RESULTS
CLU facilitates LC3 conversion
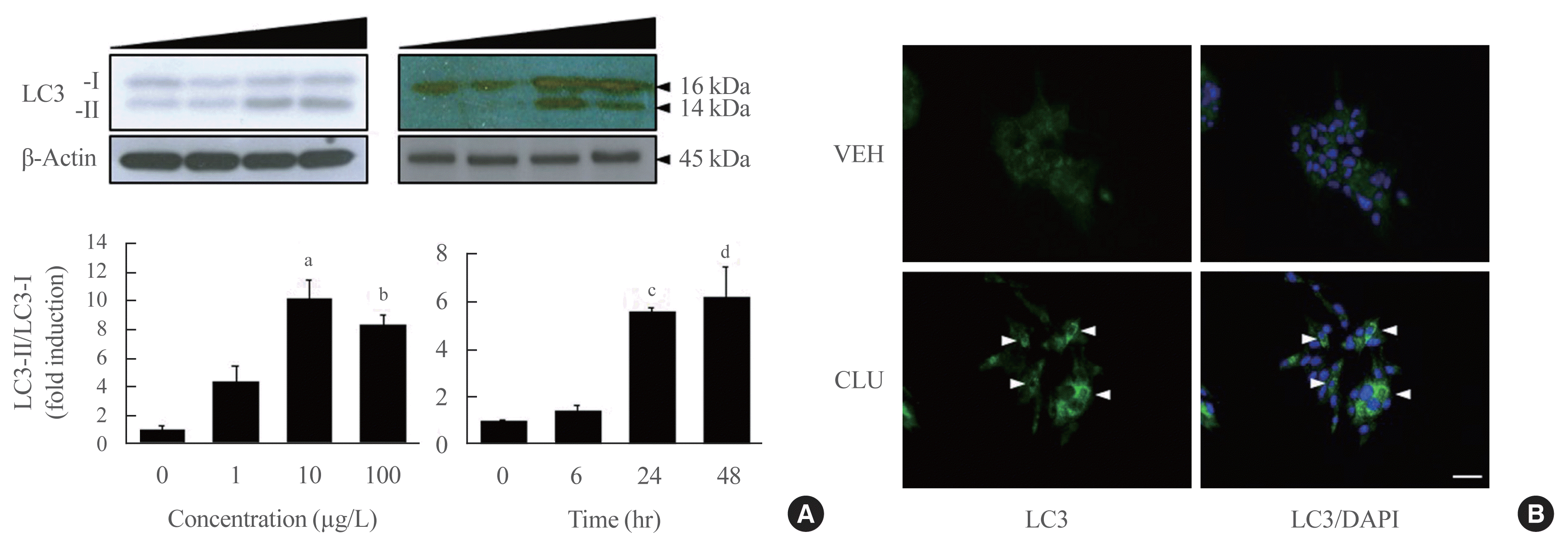
Conversion to LC3-II from LC3-I is a key step to forming the autophagosomal inner membrane in the early stage of autophagy. To examine the effect of CLU on LC3 conversion in pancreatic β-cells, MIN6 cells were treated with various concentrations of CLU at different time periods. The conversion of LC3-I to LC3-II significantly increased at 24 hours after treatment with 10 μg/L CLU (Fig. 1A). Furthermore, the fluorescence intensity of detected LC3 proteins dramatically increased after CLU treatment compared to vehicle-treated cells (arrowhead in Fig. 1B), indicating that CLU treatment increases LC3 protein levels in pancreatic β-cells.
CLU increases LC3 protein binding with Atg3 and p62
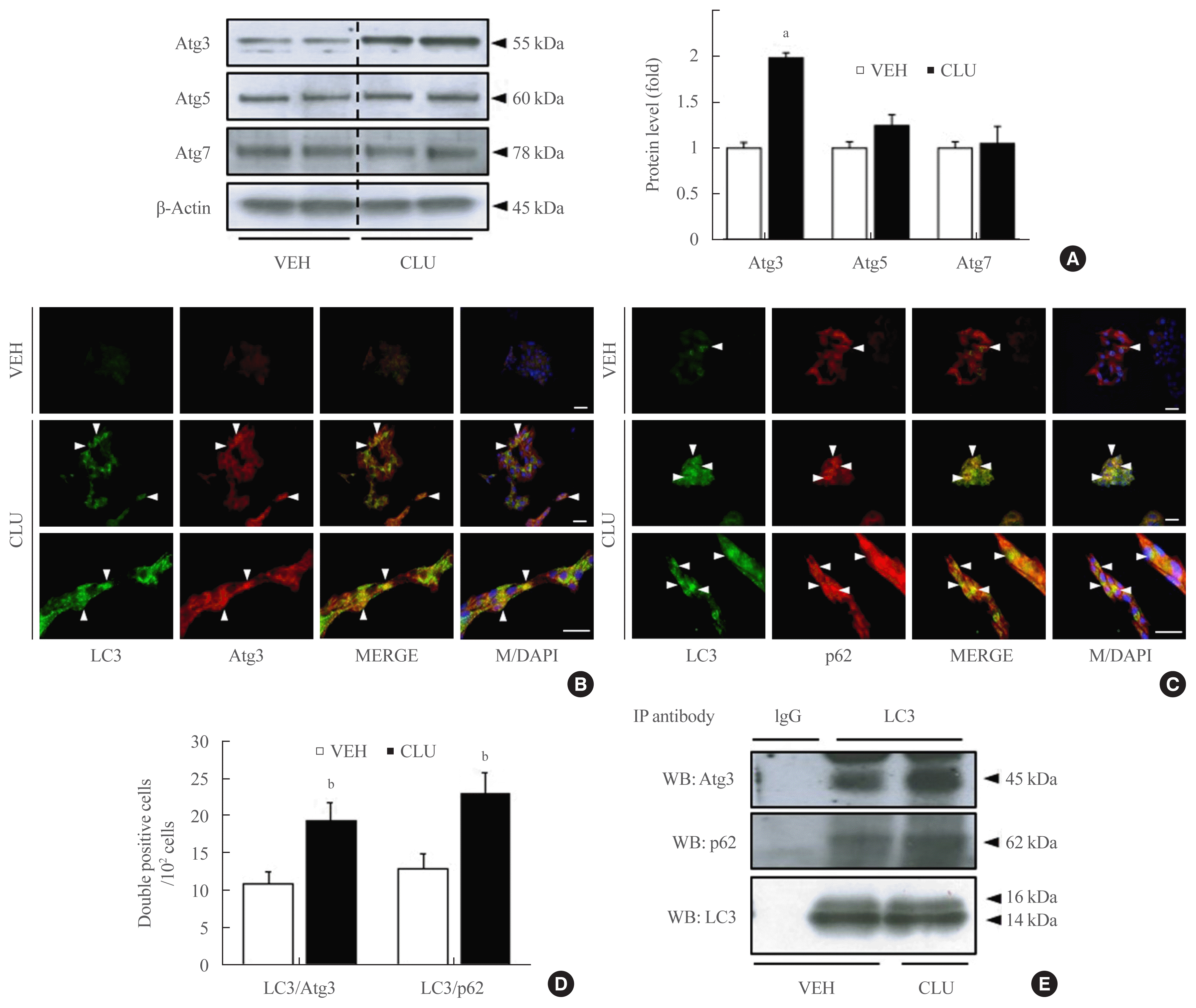
LC3-II conversion is modulated by the Atg family of proteins. In particular, Atg3, Atg4, and Atg7 directly combine with LC3-I, leading to LC3-II conversion. To define how CLU facilitates LC3-II conversion, we examined the expression levels of Atg proteins. Treatment with CLU selectively increased the protein level of Atg3 but not the other Atg proteins (Fig. 1A). In process of LC3-II conversion, Atg3 is coupled with LC3-I, thereby inducing the molecular combination of LC3-I and PE for LC3-II synthesis. We performed double labeling of the LC3 and Atg3 proteins to observe the co-localization of LC3 and Atg3. Consistent with the increase in Atg3 protein expression, the immunofluorescence signal for Atg3 was markedly enhanced by CLU, and the number of double-positive cells that simultaneously expressed both LC3 and Atg3 was significantly increased in CLU-treated MIN6 cells (Fig. 2B, D). Moreover, treatment with CLU remarkably increased the co-localization of LC3 and p62 (Fig. 2C, D). p62, an adaptor protein, binds to ubiquitinated proteins and LC3-II to accumulate the autophagy substrates via localization into an autophagic compartment, which transports ubiquitinated proteins and organelles for degradation. Next, we tested whether CLU affects LC3 binding affinity with Atg3 and p62. A co-immunoprecipitation (co-IP) assay using the LC3 antibody showed that CLU increases LC3 interaction with both Atg3 and p62 (Fig. 2E), which was consistent with the immunofluorescence image describing the overlap of LC3 with Atg3 and p62. These data suggest that CLU supports the autophagy process by facilitating LC3 interaction with Atg3 and p62.
CLU improves β-cells against free fatty acid-induced cell death and dysfunction
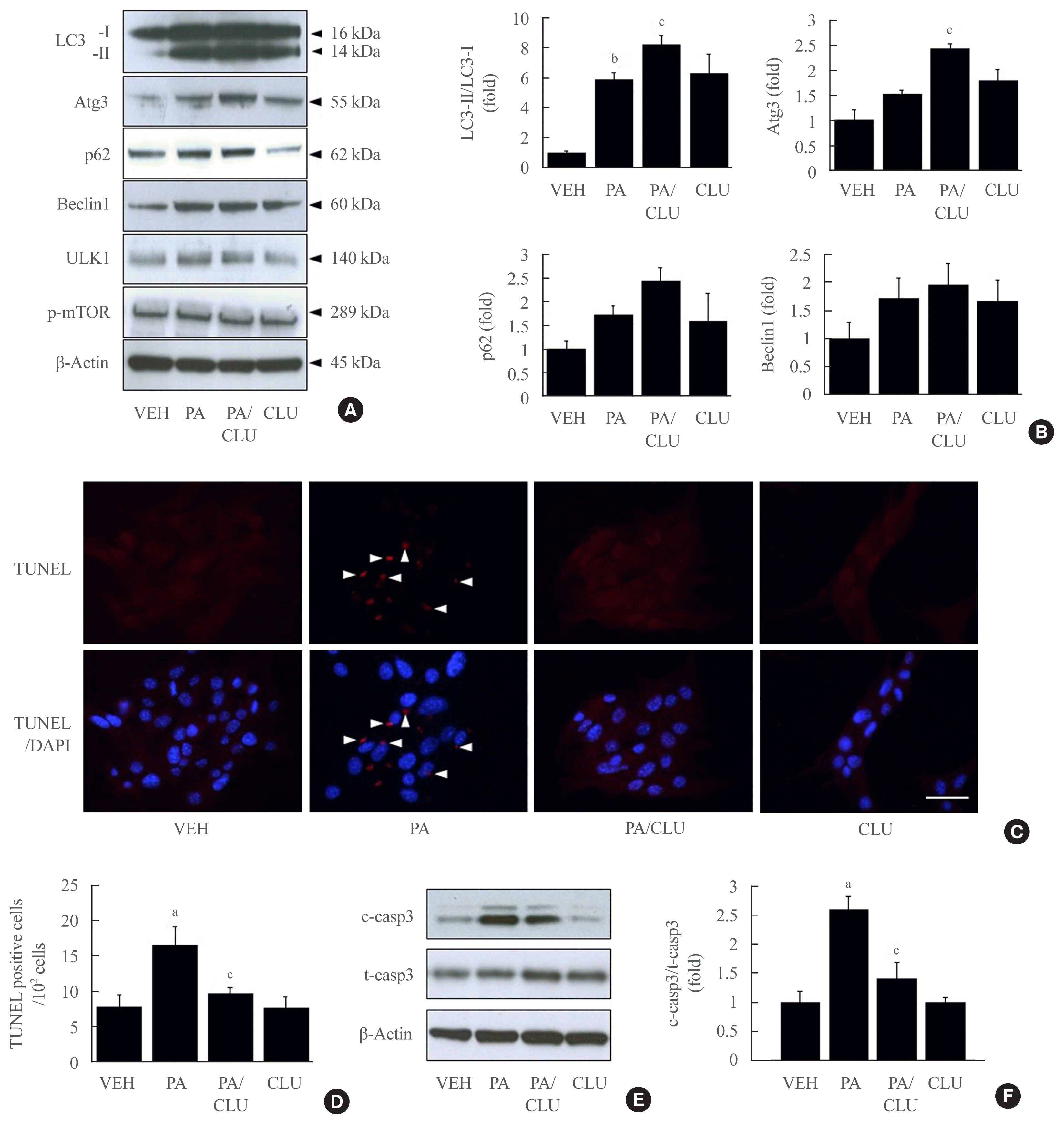
In this study, we also confirmed the role of the CLU-induced LC3-II increase in free fatty acid (FFA)-challenged β-cells. MIN6 cells were pretreated with 10 μg/L CLU or vehicle for 8 hours and subsequently exposed to 0.5 mM palmitate and/or 10 μg/L CLU for an additional 24 hours. Treatment with palmitate increased basal autophagic activity as evidenced by increased LC3-II conversion and expression of autophagy-related factors, such as Atg3, p62, beclin-1, and ULK1. Furthermore, in the cells treated with palmitate, CLU significantly increased LC3-II and Atg3 expression levels but had no effect on beclin-1 and ULK1, which are upstream regulators of autophagy (Fig. 3A, B). Next, to determine whether the CLU-induced increases in LC3-II and Atg3 contributed to the cell protection and improvement, we observed apoptosis and glucose-stimulated insulin secretion (GSIS) in MIN6 cells. Palmitate-induced apoptosis significantly decreased following CLU treatment in MIN6 cells (Fig. 3C, D), which displayed decreases in the number and intensity of terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL) positive signals (arrows in Fig. 3C). Suppression of apoptosis by CLU was confirmed with the measurement of caspase-3 cleavage. Treatment with CLU clearly reduced the cleaved caspase-3 expression level (Fig. 3E, F), indicating that CLU protects MIN6 cells from palmitate-induced apoptosis. Moreover, CLU improved glucose-stimulated secretion. Treatment with CLU significantly rescued the palmitate-induced decrease of insulin secretion in 4.5 g/L glucose-contained media (14.64 ng/mL vs. 22.71 ng/mL, P<0.05) (Supplemental Fig. S1).
Atg3 gene knockdown attenuated the protective effect of CLU
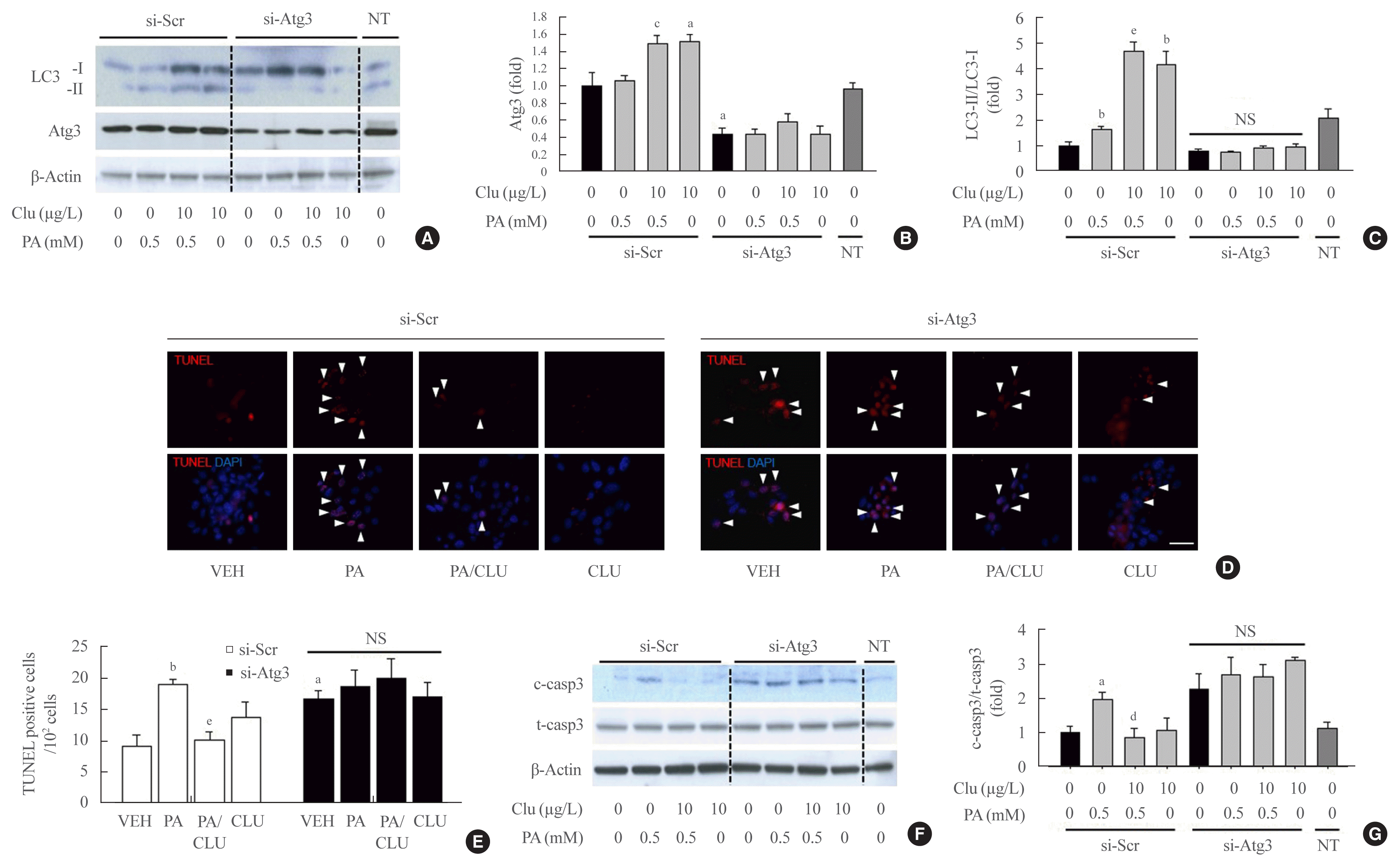
Finally, we examined whether autophagy facilitation by CLU is involved in its protective effect against lipotoxicity-induced apoptosis. Atg3 is an essential factor in LC3-II conversion and was markedly increased by CLU treatment. Therefore, we performed Atg3 gene knockdown using Atg3-specific siRNA and evaluated the effect of CLU on autophagy and apoptosis of MIN6 cells. Transfection of si-Atg3 reduced Atg3 mRNA expression to 13.6% (Supplemental Fig. S2) and protein expression to 37.3%. Down-regulation of Atg3 significantly attenuated the CLU-induced facilitation of LC3-II conversion (Fig. 4A–C), meaning repression of autophagy. In addition, knockdown of the Atg3 gene simultaneously eliminated the CLU-induced cytoprotective effect. Lipotoxicity-induced apoptosis, detected with a red signal in the TUNEL assay, was not recovered by CLU in si-Atg3-transfected cells and was also significantly decreased in si-scramble (Scr)-treated cells (Fig. 4D, E). Furthermore, Atg3 gene knockdown reversed the inhibition of caspase-3 cleavage by CLU (Fig. 4F, G). These data suggest that autophagy is a pivotal mediating process for CLU-induced cell protection against lipotoxicity in pancreatic β-cells.
DISCUSSION
Several recent studies have suggested that CLU is an important factor for the functional operation of autophagy. It has been reported that CLU expression was required to activate pro-survival autophagy in kidney tubular epithelial cells [23]. Autophagy activation by CLU was also observed to enhance cancer cell survival [24], meaning that the cytoprotective effect of CLU could be due to its role in autophagy facilitation.
In the present study, we described how CLU protects pancreatic β-cells against lipotoxicity-induced apoptosis via autophagy activation and discovered that CLU plays a pivotal role in autophagy facilitation in pancreatic β-cells. Treatment with CLU significantly increased Atg3 expression and co-localization with LC3 as well as binding between Atg3 and LC3, thereby facilitating LC3-II conversion. Moreover, CLU remarkably suppressed TUNEL signaling and caspase-3 cleavage, and knockdown of the Atg3 gene attenuated CLU-induced cell prevention, suggesting that upregulation of autophagy and Atg3 expression mediates the cytoprotective effect of CLU against lipotoxicity-induced apoptosis in MIN6 cells.
It is still unclear whether autophagy activation caused by lipotoxicity in pancreatic β-cells is a pro-survival or pro-death mechanism. However, recent studies suggest that autophagy activation is a cell protection mechanism, not a programmed cell death mechanism [8,23–26]. In particular, autophagy in pancreatic β-cells has been considered as a defense mechanism against various type of cell damage though the disposal of toxic aggregates [8,25,26]. It is conceivable that pancreatic β-cells place a high burden on protein synthesis and folding due to their role in insulin production, and the autophagy system plays a key function in resolving this burden. In the present study, CLU upregulated autophagosome formation through an increase in LC3-II conversion. Furthermore, the TUNEL assay revealed that autophagy activation by CLU markedly reduced FFA-induced apoptosis. These findings suggest that β-cell impairment by lipotoxicity is characterized by the accumulation of protein aggregates and that autophagy activation can be an expedient mechanism to reduce this impairment.
The present study also revealed that CLU enhances LC3-II conversion by increasing Atg3 expression and the binding affinity between LC3 and Atg3. LC3-II, a lipidated form that is converted from LC3, is an essential contributor to the elongation of autophagosomes and thus serves as a widely accepted marker of autophagy [27]. During the process of conversion from LC3 to LC3-II, various Atg proteins serve as catalytic enzymes or temporary binding ligands. Atg4 cleaves LC3 to generate LC3-I, and the Atg7 homodimer, the Atg12-Atg5-Atg16L1 complex, and Atg3 are sequentially involved in LC3-II conversion. Atg3 dissociates LC3-I from the other Atg proteins and subsequently promotes LC3-I/PE conjugation, leading to the generation of functional LC3-II for autophagosome elongation [28]. Firstly, we showed a remarkable increase in protein level of Atg3 by CLU treatment (Fig. 2A), and this is consistent with data of Zhang et al. [24] showing that Atg3 expression was down-regulated in heart tissues of CLU gene knockout mice. These results suggest that CLU is involved in regulation of Atg3 expression. We next demonstrated that CLU enhanced the binding between Atg3 and LC3 using co-IP. CLU is known as a chaperone, which roles an integral part of protein quality-control system regulating recognition and disposal of misfolded proteins. CLU contains amphipathic and coiled-coil helices in addition to large intrinsic disordered regions, and these structure lead to bind CLU with various proteins as well as regulates its function. Indeed, CLU enhanced proteasomal degradation I-κB by interaction with SCF-βTrCP E3 ligase [29], and regulated the apoptosis via interaction with Bcl-x(L) [30]. In the process of LC3-II conversion, Atg3 roles as an E2-like enzyme that conjugates LC3-I with PE [31]. CLU might facilitate the Atg3 function via interaction with Atg3.
A pivotal finding of the present study is that CLU displays direct involvement in LC3-II conversion via the facilitation of binding between Atg3 and LC3 without altering the upstream pathway of autophagy. Upstream regulators like beclin-1, ULK1, and phosphorylated mTOR were not altered by CLU, and moreover, lack of Atg3 resulted in a significant reversal of the CLU-induced cytoprotective effect. Therefore, CLU is a specific autophagy agonist that can activate autophagy without impacting other signaling systems in pancreatic β-cells.
CLU is a molecular chaperone that inhibits physical and chemical stress-induced aggregation of intracellular proteins and serves as a protective molecule by combating cellular impairment in numerous tissues and cells [17,18,32]. The protective effect of CLU has been principally reported in several studies about cancer and ischemic diseases. Many reports have demonstrated that CLU confers resistance to anti-cancer agents in various cancers [33,34], and the role of CLU in autophagy has been shown in studies using cancer cells [23]. Alnasser et al. [23] also reported that CLU plays a role in autophagy-mediated cell protection against hypoxia-mediated injury in kidney epithelial cells. Meanwhile, in the pancreas, it has been found that CLU was upregulated in pancreatic islet and acinar cells upon injury by streptozotocin injection or FFA treatment and served to induced differentiation among insulin-secreting cells during pancreatic regeneration [35–37]. A number of studies have reported on the protective effect of CLU in pancreatic cells [35,38], but the exact mechanism remains unclear. Here, we discovered that CLU facilitates autophagy in pancreatic β-cells, which will be helpful for future CLU-related studies of the mechanisms of β-cell protection in relation to diabetes treatment.
In conclusion, autophagy regulation is an essential component of β-cell protection against FFA-induced cell death. Thus, as a facilitator of autophagy, CLU may be relevant to the management of FFA-induced diabetes mellitus.
 XML Download
XML Download