

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Only a few grams of glucose (i.e., the plasma glucose pool size, 100 mg/dL blood or 3 g for a 70 kg healthy person with a plasma volume of 3 L) circulate in the blood, which supplies all the necessary fuels (i.e., glucose) to critical organs such as the brain. Accordingly, the maintenance of the infinitesimal size of this plasma glucose pool, a physiological priority, is typically stable in healthy individuals, even in perturbed situations such as during exercise [1–7] or after a meal intake [8,9]. For example, even following a 75 g glucose challenge (25 times the plasma pool size, similar to the amount of sugar contained in two regular Cokes), the plasma glucose concentrations typically do not rise above 200 mg/dL (<2-fold the pool size) and then return to the baseline within 2 hours in healthy individuals [8,10], as opposed to the insulin-resistant state [11] or diabetic conditions [12,13]. The tight regulation of blood glucose concentrations is achieved through the cooperative interplay among multiple organs including the brain, liver, pancreas, muscle, and adipose tissue at multiple levels of regulation [9,14]. Regardless of the complexity of control network for glucose metabolism, however, the plasma glucose pool size is regulated through changes in the rate of glucose appearance (Ra glucose) into blood from all sources and the rate of glucose disappearance or disposal (Rd glucose, or peripheral glucose uptake) from the blood [15,16]. Thus, the plasma glucose concentrations are constant as long as there is a close match between two rates, regardless of their actual rates. However, a persistent imbalance between Ra and Rd glucose leads to a fall (Ra<Rd) or a rise (Ra>Rd) in the plasma glucose pool size or concentrations (if we assume that total plasma volume is constant), ultimately leading to clinical conditions including diabetes, coma, and even death [17,18]. Changes in the plasma glucose pool size or concentrations do not reveal the dynamic status of glucose metabolism (i.e., metabolic flux). Therefore, it is crucial to obtain dynamic information (i.e., metabolic flux or kinetics) for elucidating the origins of metabolic dysregulation. The determination of metabolic flux can be accomplished with the use of stable isotope tracer methodology, which can be used in conjunction with traditional tools of cellular and molecular biology for an in-depth understanding of metabolism. In this review, we will discuss the following: (1) the importance of obtaining dynamics (i.e., metabolic kinetics) of glucose metabolism; (2) basic principles of stable isotope tracer methodology; (3) calculations of selected in vivo glucose kinetics in steady- and non-steady states; and (4) biological applications of assessments of metabolic flux in vivo (in mice) and in vitro (in cell lines).
Dynamics vs. static, snapshot of metabolism: why does it matter?
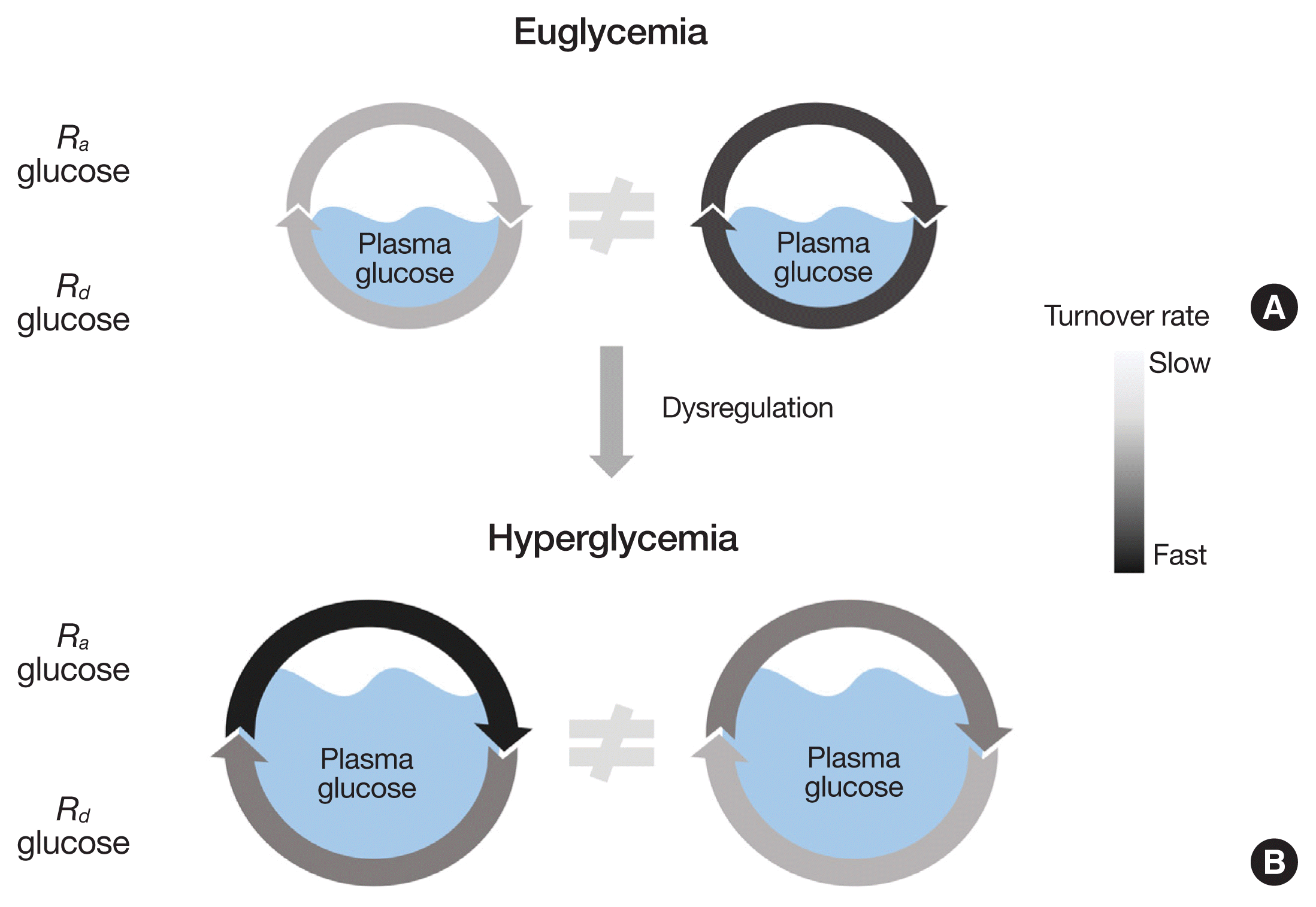
An intuitive representation of the importance of obtaining kinetic information rather than simply depending on “snapshot” or static information for a better understanding of metabolic status is depicted in Fig. 1. From the glucose pool size (euglycemia or hyperglycemia), the dynamic status of glucose metabolism (i.e., rates of glucose turnover) cannot be envisioned as it was demonstrated that the rate of glucose turnover varies by 4-fold for a given pool size [19]. Numerous permutations of rates of Ra and Rd glucose could lead to an identical pool size of glucose in the blood. Despite the dynamic nature of glucose metabolism, metabolic research heavily relies on measurements of static information such as abundance of mRNA, protein, or metabolites, or the activity of cellular signaling cascades regarding metabolic flux rates [20]. While worthwhile, the sole dependence on static, or “snapshot” information regarding the organism’s metabolic status can lead to erroneous conclusions on the actual metabolic flux rates. Regarding this notion, numerous examples have been reported in animals [20,21] and humans [22,23]. For example, the duration of fasting time is known to be positively related to the magnitude of gluconeogenesis (GNG) in the liver [21]. In accordance with this notion, it was demonstrated that fasting for over 48 hours in rats showed significant elevations in phosphoenolpyruvate carboxykinase (PEPCK), the enzyme for the first committed step in GNG in the liver [21]. However, the corresponding in vivo flux rate (i.e., GNG) was actually reduced compared to control and 8-hour fasting conditions [21]. This mismatch is not limited to the case of glucose metabolism. Neinast et al. [20] reported that pharmacological inhibition of branched chain α-keto acid dehydrogenase kinase resulted in an activation of branched chain α-keto acid dehydrogenase, the rate-limiting enzyme for the oxidation of branched chain amino acids (BCAA) without corresponding elevations in the rate of BCAA oxidation. Furthermore, in the field of human muscle protein metabolism, it has often been reported that changes in muscle protein synthetic response to anabolic stimuli such as protein/amino acids and/or resistance exercise are not associated with the activation of implicated signaling molecules (e.g., the mechanistic target of rapamycin complex 1 signaling axis) [23,24]. Discrepancies between static information (e.g., enzyme abundance/activity) and actual metabolic flux rates for a given metabolic pathway are due to the fact that the metabolic flux rates are determined by multiple factors including enzyme activity, availability of substrate, and implicated signaling [25]. While it is clear that determining metabolic flux is of critical importance for an understanding of metabolic status in addition to obtaining the traditional static information, in the following section, we will discuss basic principles of stable isotope tracer methodology.
BASIC PRINCIPLES OF STABLE ISOTOPE TRACER METHODOLOGY
Dynamic information on the metabolic status of substrates including glucose, lipids, amino acids/proteins in organisms (i.e., metabolic flux or kinetics) in vivo and in vitro can be best explored with the introduction of one or more of molecules that are labeled with either a stable or radioactive isotope (i.e., metabolic tracer) into the circulation in the body [11,15,16,26–28]. Here our emphasis will be placed on the applications of stable isotope tracer methodology owing mainly to (1) safety issues, particularly for humans and (2) the versatility of stable isotope tracers in assessing various aspects of metabolism [7,15,16,29–35]. In addition, assessments of metabolic flux using stable isotope tracer methodology are typically accomplished in conjunction with the use of gas or liquid chromatography mass spectrometry (GC- or LC-MS) [36,37] or magnetic resonance spectroscopy [38,39] with essentially the same purpose: i.e., determining tracer enrichment (e.g., magnitude of labeling, expressed as various ways) [15,16], which will be briefly discussed below. In this review, our discussion will be based on the use of GC-MS or LC-MS. In this section, we will briefly discuss (1) the meaning of a stable isotope tracer and its enrichment; (2) two basic tracer models for assessing various aspects of metabolic kinetics; and (3) calculations of glucose kinetics in different biological states. More comprehensive and detailed information is available elsewhere [15,16,40–43].
Stable isotope tracers
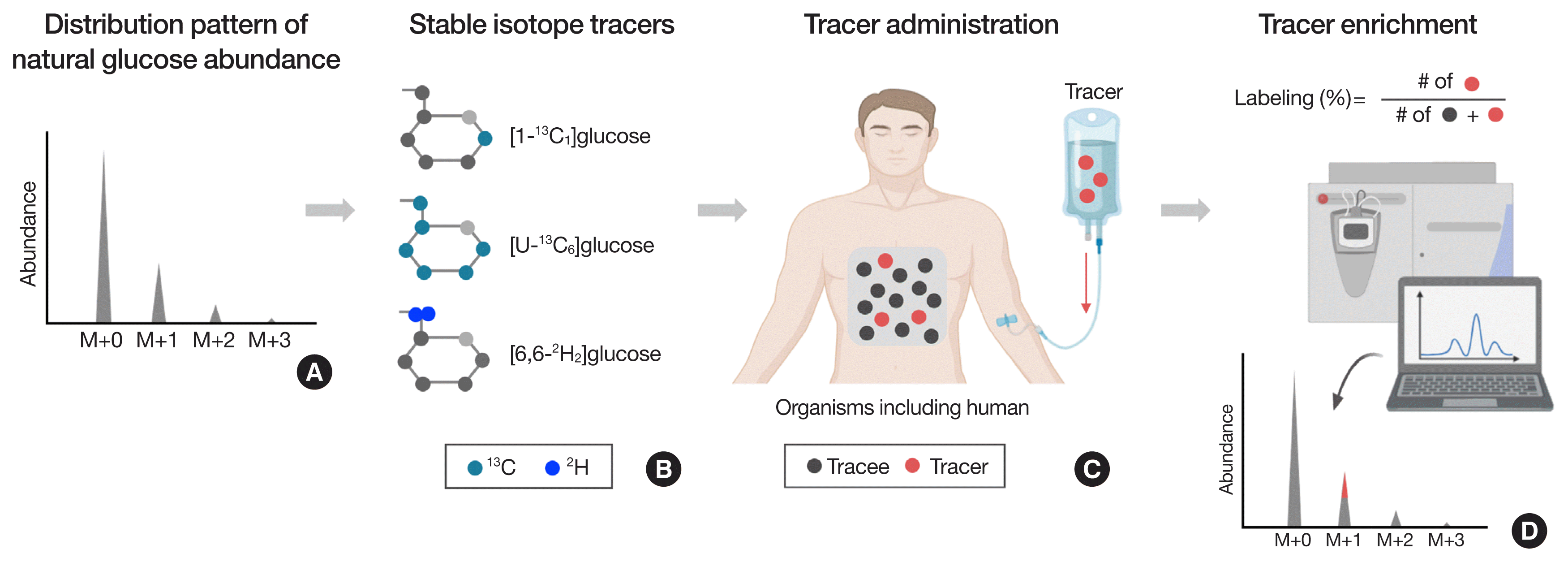
Glucose occurs naturally in varying masses with a distinct distribution pattern due to the existence of varying numbers (M+0, M+1, M+2, …) of heavier isotopes (e.g., 13C, 2H, 17O, and/or 18O) incorporated in the molecule (Fig. 2A). The most abundant mass of glucose is the lowest one (M+0; 180 molecular weight [MW]) consisting only of the lowest mass of the respective elements (i.e., 6×12C+12×1H+6×16O=180 MW). Any glucose molecule heavier than M+0 can be used as a metabolic tracer for tracing the metabolism of an endogenous tracee (e.g., glucose) and related metabolic pathways. Fig. 2B shows several examples of glucose tracers labeled with 13C at the carbon 1 position ([1-13C]glucose) or all carbon positions ([U-13C6]glucose; uniformly [U]) or labeled with two deuterium atoms at the carbon 6 position (i.e., [6,6-2H2]glucose). The selection of ideal tracer(s) is of critical importance for tracing the specific metabolic fates of the tracee of interest [15].
Enrichment: relative tracer abundance
To follow the fate of the tracee of interest (e.g., glucose), one or more of tracer is administered in different ways such as a bolus injection orally or intraperitonially (IP), constant infusion intravenously (IV), or in combinations into a body compartment (Fig. 2C) [15,16,40]. The next step is to determine tracer enrichment in the compartment when achieving a targeted tracer enrichment. Tracer enrichment is defined as the abundance of tracer relative to the tracee. There are several ways of expressing tracer enrichment, including the TTR and MPE: TTR is the tracer-to-tracee ratio (i.e., the ratio of the number of red circles to the number of black circles), while MPE is mole percent excess (i.e., the ratio of the number of red circles to the sum of all circles, % labeling) (Fig. 2D). To determine tracer enrichment (either TTR or MPE), the abundance of varying masses of the compound of interest needs to be analyzed by instruments such as GC-MS or LC-MS. In this review, we will use MPE (i.e., % labeling) for assessing glucose kinetics in vivo and in vitro.
Basic models of tracer methodology
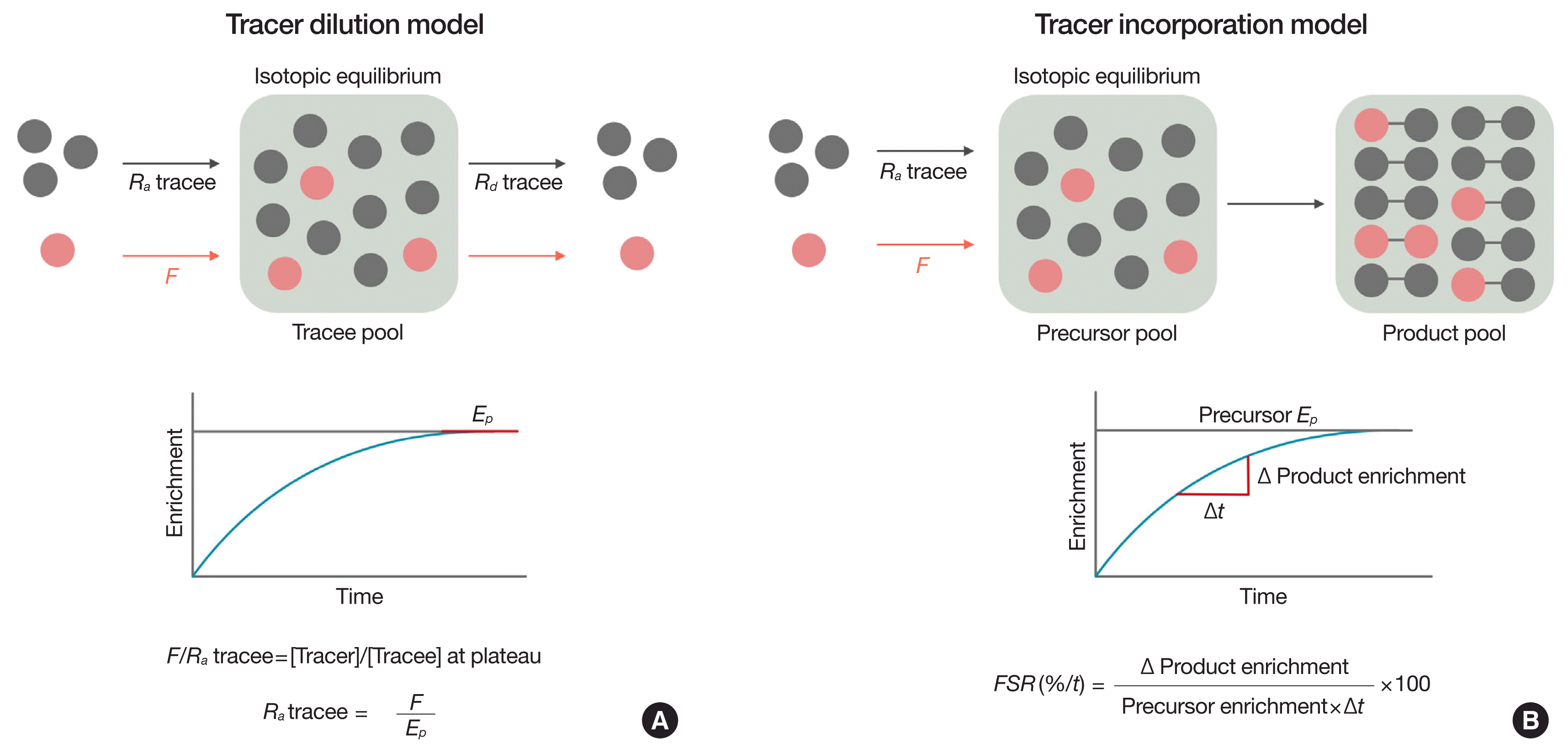
In a general sense, there are two basic models of tracer methodology to calculate tracee kinetics: (1) the tracer dilution model and (2) the tracer incorporation model (Fig. 3). While the tracer dilution model can be used alone, the tracer incorporation model is used in conjunction with the tracer dilution model in assessing tracee kinetics (e.g., GNG and glucose oxidation). Here we will briefly describe each model in physiological steady states. Non-steady state calculations are more complex and discussed briefly later in this review.
Tracer dilution model
A schematic representation of the principles of the tracer dilution model is depicted in Fig. 3A. To determine Ra tracee (e.g., glucose), a tracer of the same compound (e.g., [6,6-2H2]glucose tracer) is constantly infused into the blood at a predefined rate (F), typically after administration of a priming dose of the same glucose tracer (called a primed constant infusion). The practical purpose of administering a priming dose is to reduce the time needed to reach to the point at which tracer enrichment is constant, referred to as plateau enrichment (Ep), while not affecting the magnitude of enrichment at isotopic equilibrium (i.e., Ep). When a tracer is constantly infused into the blood at a given F, tracer enrichment will rise and eventually reach Ep, the magnitude of which is inversely related to the turnover rate of the tracee of interest for a given F. At physiological steady states where the pool size is constant, Ra tracee is equal to Rd tracee. At Ep, Ra tracer (i.e., F) is also equal to Rd tracer. The relation between Ra tracer (i.e., F) and Ra tracee dictates the tracer-to-tracee ratio at Ep. Thus, the following relations can be established: F:Ra glucose=[tracer]:[tracee]. After rearrangement of the equation, we can see that Ra glucose is the product of F and 1/MPE, which is then equal to F/Ep. From this relation, it is clear that the magnitude of dilution of the tracer by the tracee in the compartment at Ep directly reflects Ra tracee. In other words, the faster the tracee turnover, the lower the Ep for a given F. The tracer dilution model can be applicable to assessments of in vivo kinetics of various metabolites including amino acids [26,44–46], palmitate [6,47,48], glycerol [49–51], pyruvate [52], lactate [53–55] as well as glucose [8,11,56,57] in steady states and non-steady states.
Tracer incorporation model
Alterations in rates of polymer synthesis such as protein [26,58], DNA [59,60], RNA [59,61], triacylglycerol [62,63], and glucose (e.g., GNG) [64,65], from their respective precursors have clinical implications [66,67]. In this regard, the tracer incorporation model has been widely used to obtain the polymer synthesis rate and is predicated on the rate of tracer precursor incorporation to product for a given time period, yielding the fractional synthesis rate (FSR), which refers to the percent of the pool size that has been newly synthesized from the precursor for the most recent defined time period (%/time) [68]. Fig. 3B represents the schematic principles of the tracer incorporation model to determine polymer FSR. To assess FSR, various precursor tracers can be infused. After achieving isotopic equilibrium of a precursor enrichment (i.e., Ep), labeled and unlabeled precursors will be incorporated into a product (made of two identical monomers, like the synthesis of glucose from two 3-carbon precursors) in proportion to their relative abundance (i.e., precursor enrichment). Thus, the magnitude of product enrichment will be determined by (1) the precursor enrichment at Ep and (2) the rate of product synthesis [69]. FSR is calculated as the change in product enrichment divided by precursor enrichment (assumed to be constant) multiplied by 100 (for the expression as %/time) for a given time period. As its definition implies, FSR is a relative term, denoting the % of the product pool size that has been newly synthesized in the most recent time period. Thus, to obtain the absolute synthesis rate, the FSR term must be multiplied by the pool size (or the production rate of the polymer product when it is released into other compartments, like glucose). The importance of obtaining absolute rates becomes clear when comparing FSRs between two distinct pool sizes (one being half of the other). In this case, an identical muscle protein FSR obtained from two heterogenous groups translates to 2-fold differences in the absolute muscle protein synthesis rate. If one can assume the pool size is similar between groups or constant over the experimental time period, then the simple comparison of FSRs is valid [70,71]. The incorporation model can be applicable to the assessments of the kinetic calculations of various compounds, including the FSR of individual proteins [72,73] and subgroups of proteins (mitochondrial, sarcoplasmic, and mixed muscle protein) [74–79], the substrate oxidation rate [6,80–82], the DNA synthesis rate (thus cell division) [59,60], the nitric oxide synthesis rate from its precursor arginine [83–86], and GNG [30,87], the latter of which will be discussed in the following section in detail.
CALCULATIONS OF IN VIVO GLUCOSE KINETICS
Overview of glucose metabolism
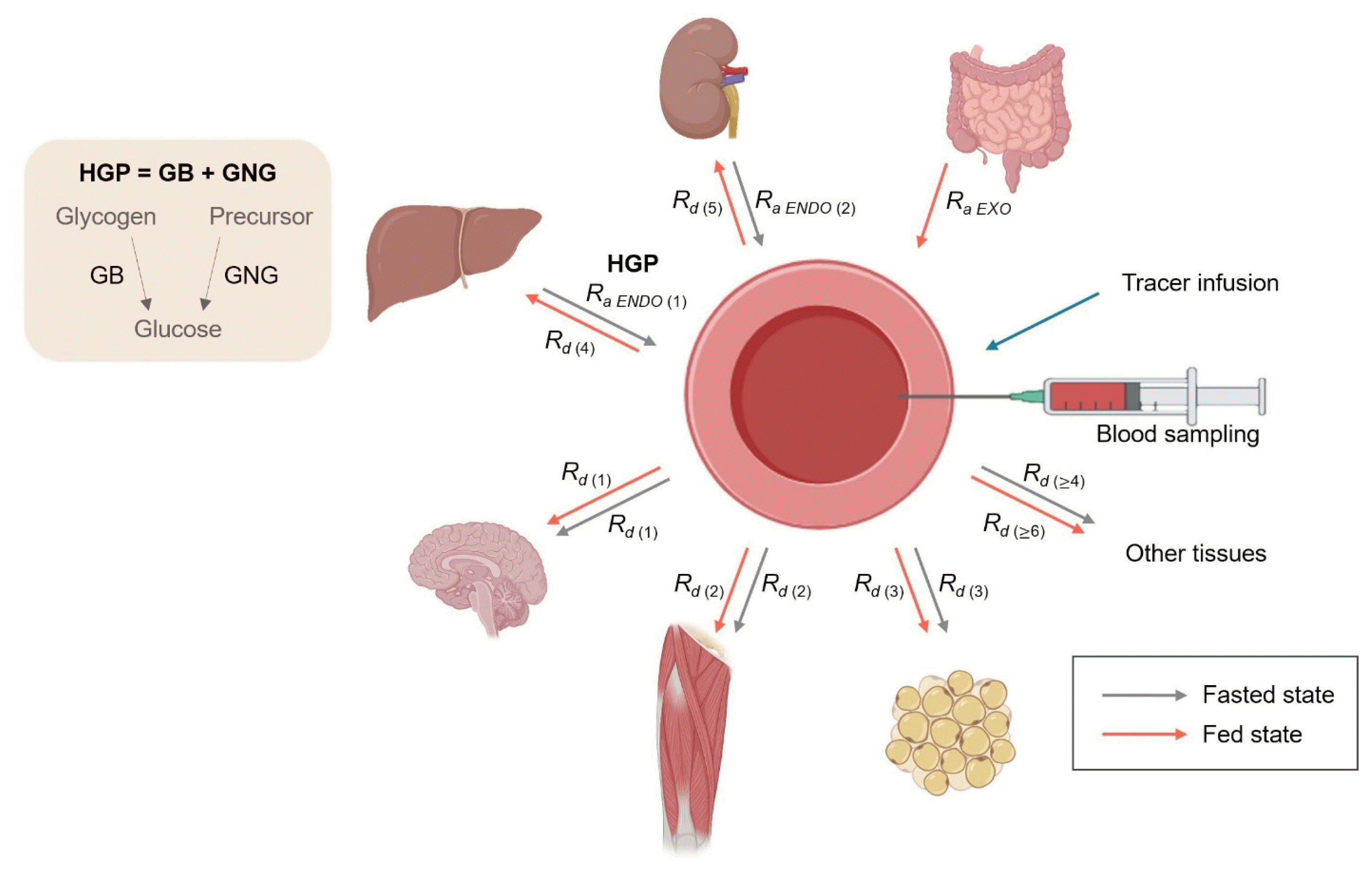
A schematic of glucose metabolism from the metabolic flux point of view is depicted (Fig. 4). In this section, the calculations of selected kinetics of in vivo glucose metabolism, centered around systemic glucose kinetics such as hepatic glucose production (HGP) and peripheral glucose uptake (i.e., Rd glucose), will be covered in steady state and non-steady state conditions (Fig. 4). In the fasted state, there is apparently no glucose appearing from the gut; instead, it appears endogenously. Endogenous glucose production (EGP) is reflected as Ra glucose in the fasted state, determined as the F of the glucose tracer divided by the Ep of glucose in the plasma. EGP consists of HGP and renal GNG in the fasted states. Further, HGP consists of (1) hepatic glycogenolysis (GB) and (2) hepatic GNG, and contributes most of the EGP. The kidneys contribute to EGP to a very minor extent only through GNG (approximately 5% of HGP unless in conditions of long-term fasting) [88]. Ignoring renal GNG due to its minimal contribution to EGP, we can assume that HGP equates to EGP. Thus, HGP can be calculated as Ra glucose. As HGP is the sum of GB and GNG, either GB or GNG needs to be determined to establish the other. It is typical to assess GNG using various tracer methodologies [64,89,90]. Thus, by knowing HGP and GNG, GB can be inferred from the difference between HGP and GNG, which will be discussed below in more detail. Because the plasma glucose pool in the basal fasted state is constant, Ra glucose is equal to Rd glucose. Rd glucose consists of the sum of the rates of glucose uptake by multiple organs/tissues (i.e.,
). To further delineate the fates of Rd glucose (i.e., identifications of specific tissues, specific intracellular metabolic pathways such as glycolysis, mitochondrial tricarboxylic acid [TCA] cycle and pentose pathways within that tissue, etc.) [42,91,92], additional techniques (e.g., deoxyglucose injection, arteriovenous balance, and tissue biopsy for intracellular metabolite labeling) [93,94] may be required. In the fed state such as following a meal intake or a test glucose dose during an oral glucose tolerance test (OGTT), the liver and kidneys become net consumers of plasma glucose. While the brain and the rest of the nervous system are the largest consumers of plasma glucose in the fasted state, skeletal muscles become the largest consumer in the fed state, accounting for up to 85% of postprandial glucose utilization [95]. In contrast to the fasted state, postprandial Ra glucose (i.e., a non-steady state) consists not only of HGP and renal GNG, but also of the contribution from the gut (i.e., Ra EXO), originally from food intake. To assess Ra EXO, a glucose tracer labeled differently from that infused IV needs to be orally consumed (e.g., [1-13C]glucose vs. [6,6-2H2]glucose), which will be discussed later in this review. Taken together, the plasma glucose is in a dynamic state of constant turnover with the cooperation of multiple organs to maintain glucose homeostasis. In the following section, we will deal with calculations of these glucose kinetics discussed above in the conditions of fasted and fed states.
Calculations of glucose kinetics in vivo in the basal fasted state
In the basal fasted state, the plasma glucose pool size is constant, implying a close match between Ra glucose (i.e., HGP) and Rd glucose. In the basal fasted state, Ra glucose (=Rd glucose) represents EGP or HGP (via GNG and GB) if ignoring renal GNG. To explore the glucose kinetics, we first need to discuss calculations of Ra glucose in the basal fasted state.
Endogenous glucose production
To determine EGP or HGP, one or more blood samples are collected for obtaining background enrichment before tracer administration. Then, after administration of a priming glucose tracer dose (a bolus injection of a glucose tracer), the same glucose tracer is constantly infused IV at a predefined F. When Ep is reached, blood samples are collected a couple of times for determination of tracer enrichment. Then, the steady state kinetics of the tracee (i.e., Ra glucose) can be calculated as F divided by Ep. For instance, if [6,6-2H2]glucose (M+2) is infused constantly at 0.80 μmol/kg/min following a priming dose of 80 μmol/kg, and if the value of Ep glucose is 0.04 (MPE=4%) after subtracting the background enrichment, then Ra glucose, calculated as 0.80 μmol/kg/min divided by 0.04, would be 20 μmol/kg/min. Because the plasma glucose pool is constant, Rd glucose must be 20 μmol/kg/min as well. As discussed above, Ra glucose represents EGP, largely reflecting HGP. As HGP is the sum of rates of both hepatic GB and GNG, by knowing GNG, GB can be inferred.
Hepatic gluconeogenesis
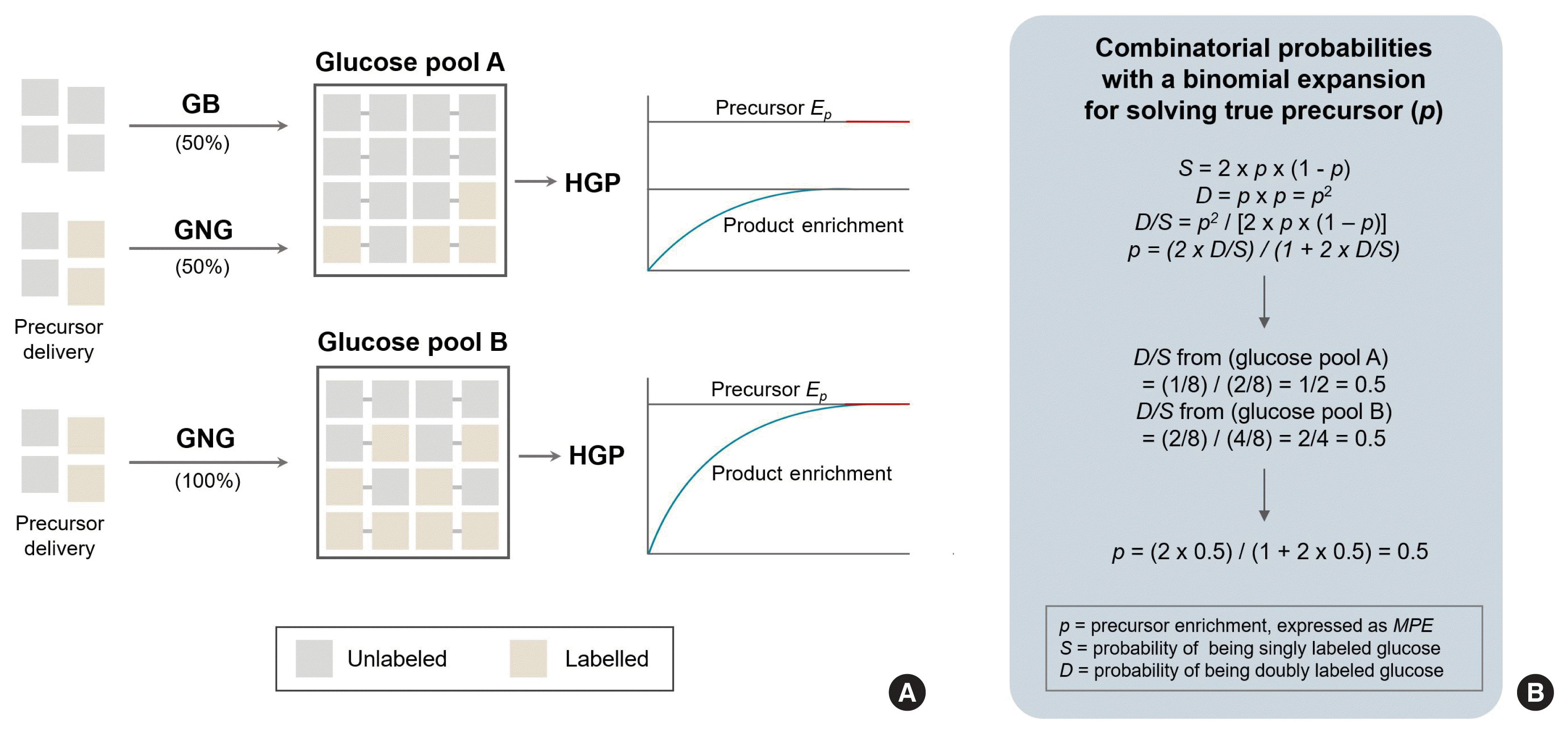
Hepatic GNG is calculated as the product of the fractional contribution of GNG to HGP (FGNG) and HGP (=Ra glucose in the fasted state). FGNG is determined in relation to the precursor (3-carbon sugar) product (i.e., glucose) rule based on the tracer incorporation model as described above. As described in the FSR calculation above, FGNG is calculated based on the relationship between the enrichment of the product and that of the precursor. While it is straightforward to identify the enrichment of the product, as new glucose appears into the circulation where plasma glucose can be sampled, determination of the “true” precursor enrichment has been a debatable issue over decades [15,96] due to a number of issues that hinder the quantification of “true” precursor enrichment [96,97]. For example, with a primed constant infusion of a precursor tracer (e.g., tracers of glycerol, pyruvate, alanine, or lactate) [51,52,55,98], it is typical to obtain precursor tracer enrichment from plasma (particularly in the case of humans) [52,99] or directly from liver tissue (typically in animal models) [100,101]. Problems arise at multiple levels including (1) dilution of tracer en route to glucose in a number of points of metabolic steps and/or (2) gradation of enrichment across liver tissue, which over- or under-estimates “true” GNG [15,102]. Regarding this issue, several solutions have been developed, which are conceptually sound and currently widely used with some practical limitations mostly arising from analytical issues [103]. Among others [102,104], mass isotopomer distribution analysis (MIDA), developed by Hellerstein and Neese [96,103], and heavy water labeling methods [105] bypass the true precursor enrichment issues. In this review, we will briefly discuss the MIDA approach for the calculation of true precursor enrichment due to space limitations. Fig. 5 shows the principle of calculating the true precursor enrichment using MIDA from the pattern of enrichment of the product (glucose) in the plasma regardless of the magnitude of ongoing GB. The principle of MIDA is relatively simple and is predicated on combinatorial probabilities with a binomial expansion: nCk×pk×(1−p)n-k where n is the number of subunits in the product, k is the number of labeled subunits, and p is the labeling probability of the precursor pool [96,103]. With an Ep of the precursor of 0.5 during a constant infusion of singly labeled precursor tracer (e.g., [1-13C]glycerol), the probabilities of new glucose being both unlabeled (both, black), singly labeled (S), or doubly labeled (D, both red) in case of no GB can be calculated using the equations with the information (for glucose, n=2, k=1 for S or k=2 for D) to be 0.25 (i.e., 25%), 0.50 (i.e., 50%), and 0.25 (i.e., 25%) respectively. However, when GB (providing unlabeled glucose to the product pool) contributes 50% to HGP, the Ep of the product (i.e., glucose) will be diluted by half. While S (2/8 for pool A vs. 4/8 for pool B) and D (1/8 for pool A vs. 2/8 for pool B) may vary depending on the magnitude of GB, the ratio of D to S (D/S) is constant (i.e., 0.5 for both) for a given precursor enrichment. True precursor enrichment can be obtained by solving the equation (i.e., D/S=p2/[2×p×(1−p)]) for p by rearranging the equation, i.e., p=(2×D/S)/(1+2×D/S). Because D/S for both cases is identical, the p values calculated from both cases are identical regardless of occurring GB. Thus, FGNG is calculated by dividing Ep product enrichment by the same Ep precursor (0.5): FGNG of 50% in Fig. 5A and 100% in Fig. 5B. Thus, the absolute rate of GNG will be the product of HGP and FGNG. Thus, 25 μmol/kg/min×0.5 (50%)= 12.5 μmol/kg/min. Thus, hepatic GP will be inferred as HGP minus GNG, which is 12.5 μmol/kg/min. In the following, we will deal with the calculation of non-steady state glucose kinetics using an example of an OGTT condition.
Calculations of non-steady state glucose kinetics: Steele equations
Unlike steady state conditions, it is more complex to calculate tracee kinetics in non-steady state conditions such as after meal intake [8,11,56] and during exercise [3,4], as both the pool size and tracer enrichment of glucose are changing. In this section, we will discuss how to calculate non-steady state glucose kinetics using an example of an OGTT condition [8,11,56]. To determine non-steady state glucose kinetics, dual glucose tracers are typically used: one for IV infusion and the other for an oral glucose challenge (e.g., 75 g glucose consisting of 3.75 g labeled glucose and 71.25 g unlabeled glucose, giving an MPE of 5%). The tracer (e.g., [6,6-2H2]glucose) for IV infusion is used to trace endogenous Ra (Ra ENDO) and Rd glucose, while the oral glucose tracer (e.g., [1-13C]glucose contained in a drink mixture with unlabeled one) that is differentially labeled than the one infused is used to trace exogenous Ra glucose (Ra EXO) from the gut (Fig. 4, red line). Ra glucose in the basal fasted states can be calculated using F of [6,6-2H2]glucose (M+2) and its Ep from blood samples before the oral glucose challenge as discussed above. Following a glucose challenge, there are decreases in M+2 enrichment (i.e., [6,6-2H2]glucose) due to the appearance of glucose (largely M+0 and M+1) from the test glucose solution that dilutes M+2 enrichment (with the assumption that the oral glucose tracer is 100% of [1-13C]glucose), while there are increases in M+1 enrichment due to the appearance into the circulation of [1-13C]glucose (M+1) from the oral glucose. In contrast to the basal fasted state, postprandial Ra glucose (Ra TOT) is divided into Ra ENDO and Ra EXO. To determine Ra EXO, the fractional contribution of Ra EXO to Ra TOT is calculated as the ratio of plasma enrichment (M+1) to drink enrichment (M+1), somewhat similar to the calculation of FGNG, and then multiplied by Ra TOT to obtain Ra EXO. Contrary to the steady state calculations, there are issues in non-steady states: changes in the pool size and isotopic enrichments that violate assumptions for the calculations of steady state kinetics, which must be accounted for in the calculation of glucose kinetics in non-steady states. Many researchers have developed methods that account for these changes in the glucose pool size and isotopic enrichments in non-steady states for calculations of glucose kinetics [106,107]. The so-called “Steele” equations derived by Steele [107] in 1959 have been most extensively used to account for the nature of non-steady state conditions. The assumptions of the Steele equations include that the compounds of interest being traced in the non-steady state are either distributed in a single pool, or can be described as if the sampled compartment represents a fraction of a single pool. Among others, glucose [1,56,108,109], free fatty acids [6,7,47], glycerol [110,111], and lactate [53,112] in the plasma are compounds that essentially meet the assumptions. The details of the equations will not be discussed here as they are beyond the scope of this review given space limitations [15].
BIOLOGICAL APPLICATIONS TO EXPERIMENTAL MODELS
The basic principles of stable isotope tracer methodology are applicable to both humans and animals in vivo, as well as to cells in vitro. In contrast to humans, much more detailed flux information can be obtained from animal models (and in vitro models) for a number of reasons including the high accessibility of tissues/organs, feasibility of invasive surgical applications, and availability of large amounts of tissue. In this section, we will briefly cover the procedures of stable isotope tracer studies in mouse models (in vivo) and cell lines (in vitro).
Tracing metabolic flux in vivo in mouse models
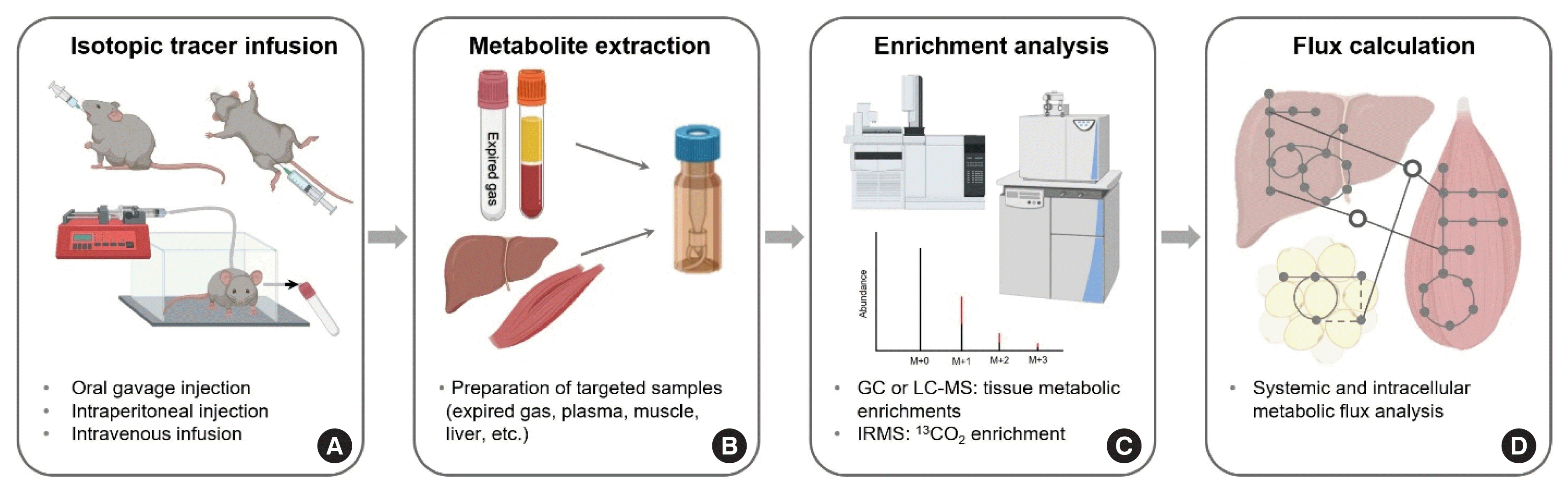
The method of non-radioactive stable isotope tracers in mouse models has been typically used to analyze whole body and tissue metabolism of a substrate of interest and other metabolites derived from the compound (e.g., glucose) due to the advantages mentioned above. Moreover, methods using a variety of tracers have been extensively utilized to study metabolic flux in a variety of clinical conditions (e.g., type 2 diabetes, cancer cachexia, and obesity) [113–115]. In the following section, we will briefly cover the essential procedures of in vivo metabolic flux studies in mouse models, including tracer administration, catheterization surgery, metabolite extraction, enrichment analysis, and flux calculation with an example of assessment of selected aspects of in vivo glucose flux (i.e., Ra glucose and oxidation) (Fig. 6).
Tracer administration and catheterization surgery
Assessment of glucose metabolism needs to be performed in the conscious state as anesthesia affects glucose homeostasis [116, 117]. Tracer administration can be achieved by using an oral gavage injection, IP injection, or IV infusion (Fig. 6A) [69,117, 118]. While useful in exploring dynamics of metabolism, both oral methods are not practically feasible for assessing absolute rates of glucose turnover due to the inability to achieve Ep and the requirement of frequent blood sampling, both of which make kinetic calculations more complicated. In contrast, a (primed) constant infusion method via IV catheterization indwelling on the right jugular vein is a frequently used technique that not only enables the determination of absolute rate of glucose turnover but also avoids excess irritation in response to gavage or IP injection [69,119]. However, the surgical procedure of catheterization requires a skillful mouse surgeon, which will be described first.
Surgical catheterization
Briefly, a median incision, approximately 5 mm, is made at a subtle palpating spot that is approximately 5 mm on the right side of the trachea, the isolation of the right jugular vein is conducted, and a silastic catheter is inserted into the vessel. The other end of the catheter is tunneled to the back of the neck for two purposes: (1) preventing mice from pulling out and damaging the catheter and (2) easy assessment of the catheter in the tracer infusion experiments. The surgical procedure can be completed in approximately 20 minutes by an experienced mouse surgeon, and it typically takes approximately 5 to 7 days for the mouse to fully recover from the surgery.
Stable isotope tracer infusion
After a 5 to 15-hour fast (depending on the research question), experimental mice undergo a (primed) constant infusion of 13C glucose. Before and at isotopic equilibrium during the tracer infusion, blood and expired air samples are collected for the calculations of systemic glucose turnover and substrate oxidation, respectively. At the end of the infusion, experimental mice are anesthetized, and tissues are harvested for determining tissue specific metabolic flux.
Metabolite extraction
Plasma and homogenized tissues are deproteinized. The supernatant containing metabolites is collected, desiccated, and chemically derivatized in order to increase the volatility and stability of metabolites at high temperature on a GC capillary column (Fig. 6B).
Enrichment analysis
Using GC-MS, the metabolites undergo the separation of substances in GC capillary columns and fragmentation of the substrate of interest. Then, tracer abundance of ion peaks will be counted using selective ion monitoring. To analyze substrate oxidation, isotope ratio mass spectrometry (IRMS) with high detection sensitivity is utilized to measure enrichment of 13CO2 from the expired air of the model animals (Fig. 6C).
Flux calculation
At the end of the procedures, various flux parameters can be calculated (Fig. 6D). First, systemic glucose flux such as Ra tracee (e.g., glucose, amino acids, fatty acids) can be calculated based on the tracer dilution model using critical information including Ep and F in fasted steady states and Ep, F, and tracee concentrations (reflection of tracee pool size) in the non-steady state. In addition, intracellular metabolite flux can be estimated via an examination of labeling patterns (i.e., patterns of mass isotopomer distribution [MID]) of each metabolite. Finally, by using 13C-labeled substrate as a tracer, the rate of substrate oxidation (e.g., rates of oxidation of glucose, palmitate, leucine, etc.) can be assessed with additional analyses of 13CO2 enrichment of expired air and CO2 production from the mice.
13C tracing in metabolic network in vitro using metabolic flux analysis
Due to its critical implications in the progression of diseases, altered intracellular metabolism has become an attractive target for the development of therapeutics. Therefore, the measurements of detailed status of intracellular metabolism has become more necessary in biomedical research. Stable isotope labeling techniques in vitro can provide intracellular metabolic flux rates in relative terms (through the examination of labeling patterns [MID] of metabolites of interest following tracer treatments) or absolute terms (with the help of computer software such as isotopomer network compartmental analysis [INCA]) [120] in controlled conditions. In this section, we will briefly cover the standardized workflow of metabolic flux analysis (MFA) using representative software (INCA) to measure the absolute rates of intracellular metabolic flux, which has emerged as the primary approach in this regard.
Metabolic flux analysis
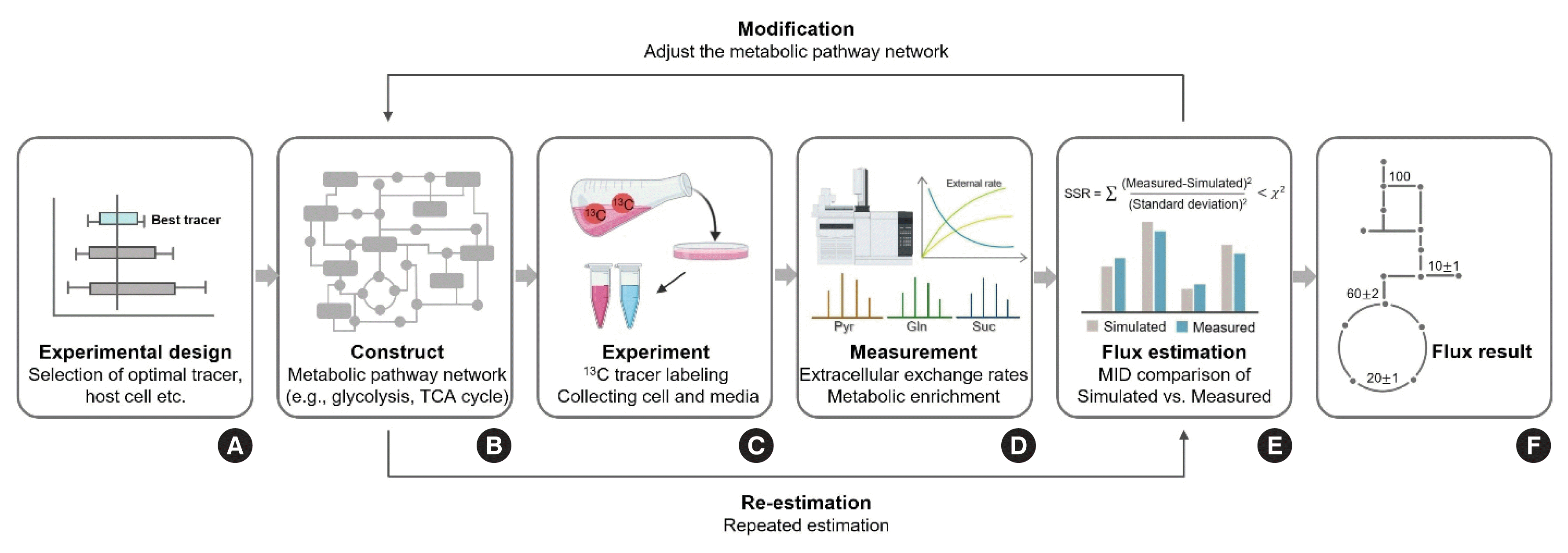
In the process of MFA with the assistance of computer software such as INCA, the labeling patterns of intracellular metabolites (i.e., MID) derived from 13C-labeled nutrient(s), cellular uptake of the nutrient(s), and secretion rates of selected product(s), and prior knowledge of the metabolic pathway network are combined for computational estimation of metabolic flux [120,121]. To examine intracellular metabolic flux, an experiment is initially designed (Fig. 7A). In the experimental design, the selection of an optimal tracer(s) is best made in silico (i.e., in computer simulations) [122] and is important to ensure an accurate analysis of metabolic flux, followed by constructing a metabolic pathway network for MFA (e.g., glycolysis pathway, pentose phosphate pathway, etc.) (Fig. 7B). A metabolic pathway network can be constructed using databases such as the Kyoto Encyclopedia of Genes and Genomes (KEGG). The next step is to measure external flux rates (e.g., rates of uptake of glucose and/or glutamine and rates of secretion of lactate and/or pyruvate) based on changes in concentrations of substrate of interest for a defined time period, normalized for the number of cells after treatment of the medium with 13C tracer(s) (Fig. 7C). Sequentially, isotopic labeling patterns (i.e., MID) of intermediates from the cells and medium are determined by using GC-MS (Fig. 7D). With the assistant of MFA software such as INCA, factors including constructed metabolic pathway network, measured external flux rates, and MIDs of intermediates are utilized for flux estimation. Then, through rigorous statistical processes, the goodness-of-fit of the constructed model to the observed values is first verified and then the sum-of-squared residuals between the simulated and measured MID results is minimized through repetitions of model reconstruction (e.g., modeling assumptions, additional information from the literature, or novel hypotheses) until being statistically acceptable (Fig. 7E) [123]. The statistically accepted results finally represent the absolute rates of intracellular metabolic flux (Fig. 7F).
CONCLUSIONS
Like other substrates, the maintenance of normal glucose metabolism is achieved through cooperative orchestrations at multiple levels of regulation including multi-omics and implicated signaling. While worthwhile, static “snapshot” information on metabolism does not reveal the dynamic nature of glucose metabolism, which is in a constant state of turnover (i.e., rates of production, appearance or disappearance, and oxidation of glucose). Stable isotope tracer methodology in metabolic studies is a state-of-the-art technology that allows both clinical and basic researchers to explore metabolic kinetics in various (patho)physiological conditions both in humans and animals in vivo and in vitro. In in vivo animal and in vitro cell studies, stable isotope tracer methodology can enable the determinations not only of systemic flux rates, but also of intracellular metabolic pathway flux rates (i.e., relative flux rates) through the close examination of MID patterns. The absolute intracellular flux rates of a specific metabolic network can also be estimated with the assistance of computer software such as INCA. Taken together, stable isotope tracers can provide an in-depth dynamic assessment of changes in metabolic status in conjunction with simultaneous investigations of the molecular basis of the observed kinetic responses.
 XML Download
XML Download