

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Thyroid cancer is relatively rare, representing approximately 3% of all new cancer cases in the United States [1]. It is, however, the most common cancer in the endocrine system, and it has had the fastest growing incidence of all cancers over the last decades [2]. Among malignant tumors affecting women, its incidence ranks fifth worldwide [3]. In the United States, about 52,900 new cases of thyroid cancer are expected to be diagnosed in 2020 [1].
While alterations on some genes and pathways have been described to be associated with the different forms of thyroid cancer [4,5], the most recent evidence indicates that the development and progression of cancer depend not only on genetic characteristics but also on the interaction of tumor cells and their surrounding microenvironment [6,7]. This tumor microenvironment (TME) is an active and dynamic component influencing cancer cell behavior, disease progression, and response to therapy in many different types of neoplasia. The TME comprises a variety of non-epithelial cells (or stromal cells). Fibroblasts are one of the main cell components of the TME [8–11].
Although fibroblasts were first described at the end of the nineteenth century [12], their role in tumor development has only recently begun to be uncovered [6]. Normal fibroblasts play an important part in tissue homeostasis and wound repair by secreting extracellular matrix (ECM) components. In non-tumor tissues they are generally quiescent. However, upon tissue damage, fibroblasts are activated to proliferate and increase their synthetic and metabolic activity. Activated fibroblasts secrete higher levels of ECM components to favor wound closure and scar formation [13,14]. Once the wound is repaired, most of these activated fibroblasts undergo apoptosis and their number decreases [15,16]. In contrast, tumors have been considered “wounds that do not heal” because the wound healing response is not self-limited and the number of activated fibroblasts is not decreased, as in normal wound repair [17]. Instead, fibroblasts in tumors, called cancer-associated fibroblasts (CAFs), are key players in tumor development [13].
Although studies analyzing the contribution of the components of the TME, particularly CAFs, in thyroid cancer progression have been limited, the thyroid TME has become an evolving field. In this review we summarize the current knowledge about the origin and markers of CAFs in thyroid cancer. We focus specifically on the recent progress in understanding how CAFs drive thyroid cancer development and progression, from the standpoint of tumor-stroma interactions. We will also discuss CAFs as a promising therapeutic target in thyroid cancer.
Go to : 
THYROID CANCER
Epidemiology and molecular pathogenesis
Thyroid carcinomas are classified as follicular-derived or C-cell-derived, according to their cell of origin. The majority of thyroid cancers originate from follicular cells, constituting approximately 95% of all cases. Follicular-derived thyroid malignancies are subdivided into differentiated thyroid cancer (DTC), poorly differentiated thyroid cancer (PDTC), and anaplastic thyroid cancer (ATC) [4,5]. DTC includes papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC). PTC is the most frequent type of thyroid cancer and is found in more than 80% of cases. It tends to grow slowly and spreads first to local lymph nodes [5,18]. FTC is the second most frequent and represents 10% to 15% of thyroid cancer cases. As with PTC, FTC can grow into lymph nodes in the neck. However, FTC is more likely than PTC to grow into blood vessels and spread to distant areas, particularly lungs and bones [19,20]. Although PDTC (1% to 15%) and ATC (<1%) are uncommon tumors, they constitute a more aggressive follicular-derived thyroid carcinoma [21–23]. Parafollicular C cell-derived thyroid cancer, which constitutes only a small fraction of thyroid cancer (approximately 2% to 5%), is classified as medullary thyroid cancer (MTC) and has distinct biological characteristics [24].
DNA sequencing studies have provided unique insights into the genetic basis for most thyroid cancers. The main oncogenic alterations of thyroid cancer occur in genes involved in mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3 kinase (PI3K) signaling pathways, such as BRAF, RAS, and RET [25,26]. The BRAFV600E mutation is the most frequent genetic mutation in PTC with a prevalence of approximately 60% [4,18]. For FTC, point mutations in RAS genes (mainly NRAS and HRAS) are the most frequent, found in 40% to 50% of the cases [25]. Activating mutations in the RET proto-oncogene account for most cases of MTC [24]. Other genetic alterations are also found in thyroid cancer, including loss of tumor protein 53 (TP53) and activating mutations in telomerase reverse transcriptase (TERT) promoter in undifferentiated thyroid cancer [21, 23,27].
Most patients with DTC have good rates of long-term survival. Standard treatment involves surgery, radioactive iodine, and thyroid hormone therapy [28]. The 5-year survival rate is approximately 98% [1]. A small percentage of DTCs, however, may manifest aggressive behavior that cannot be controlled by conventional therapy. Moreover, ATC is one of the most aggressive and lethal human malignancies, with a short median time of survival (3 to 6 months). Standard therapy was not effective in some DTC and ATC patients. Therefore, there is an urgent need to identify new biological targets that can be translated into novel clinical approaches.
Go to :
THE TUMOR MICROENVIRONMENT
The effects of oncogenic transformation of epithelial tumor cells on cancer progression have been well established. In 2000, Hanahan and Weinberg [29] proposed a list of six capabilities acquired by cancer cells during the process of malignant transformation. These six hallmarks of cancer consisted of uncontrolled proliferation, insensitivity to anti-growth signals, evasion of apoptosis, unlimited replicative potential, acquired capabilities in tissue invasion and metastasis, and sustained angiogenesis [29]. However, at that time, the heterogeneous and structurally complex nature of tumors was largely unexplored. More recently, the tumor hallmarks were updated to include TME factors that are required for malignant transformation [6,7]. Cancer cells themselves produce stimulatory factors that recruit and activate numerous normal stromal cell types from the neighboring tissues as well from the circulation. Interactions between neoplastic cancer cells and their supporting stroma create the TME, which profoundly shapes cancer progression [30]. As an example, pancreatic stellate cells (PSCs) have been reported to play a crucial role in the pathogenesis of pancreatic ductal adenocarcinoma (PDAC). PSCs represent the main cell type causing the desmoplastic reaction, which constitutes a dramatic increase in connective tissue or stromal cells that surround and infiltrate the tumor. The desmoplastic stroma contributes to tumor growth, angiogenesis, invasion, and therapeutic resistance in PDAC [31]. Thus, studies over the past two decades have revealed that the TME is an equally important hallmark of tumor behavior [7]. The TME is composed of different cellular populations, such as fibroblasts, immune and endothelial cells, among others. Fibroblasts in human tumors, also referred to as CAFs, play a key role in cancer development and progression in a variety of human tumors [8–10], including thyroid cancer.
Go to :
THYROID CANCER-ASSOCIATED FIBROBLASTS
Recruitment and activation of fibroblasts in thyroid cancer
Fibroblasts are the major cellular components of the TME. In healthy tissues, fibroblasts are quiescent, but in the TME, they are reprogrammed by the tumor to acquire tumor supporting phenotypes. Fibroblasts can be stimulated by a variety of oncogenic signals to acquire an activated state, also referred to as myofibroblasts or CAFs. Compared with quiescent fibroblasts, CAFs are distinguished by their morphology, their metabolic, synthetic, and secretory phenotypes, as well as their enhanced proliferative and migratory properties and ECM production [8–10,32]. In contrast to quiescent fibroblasts, CAFs are larger, with more cytoplasm branches. Several protein markers are used to detect CAFs, including of α-smooth muscle actin (α-SMA), platelet-derived growth factor receptor (PDGFR)-α/β, fibroblast activation protein (FAP), vimentin, and neuron glial antigen (NG2). However, most of these markers are not unique to CAFs [9], making their identification challenging in some situations.
What is known about the origin of CAFs?
Numerous studies suggest that CAFs represent a heterogeneous component of the stroma and originate from multiple types of cells. A large number of them derive from mesenchymal stem cells and host tissue fibroblasts (pre-existing resident fibroblasts), which proliferate and expand locally after stimulation caused by cancer-derived growth factors. In addition, oncogenic signals produced by tumor cells may recruit fibroblasts to the tumor site from a distant source [8,9].
What is known about the origin of thyroid CAFs?
CAFs have been found to be abnormally increased in thyroid carcinomas as compared to normal thyroid tissue. The majority of studies investigating CAFs in human thyroid cancer correlate the expression of CAF-related proteins with clinical and pathological features. In a study performed in a small sample group of patients with PTC, the presence of CAFs (defined by α-SMA staining) was associated with cervical lymph node metastasis [33]. In another study, the expression levels of different CAF-related proteins in human PTC was studied by using tissue microarrays, finding differences according to histologic subtypes and the presence of BRAFV600E mutation. Interestingly, PTCs with the BRAFV600E mutation exhibited high expression of some CAF-related proteins, such as FAP-α. This study also revealed that stromal positivity for PDGFR-β was associated with shorter overall survival in PTC [34]. In a more recent study, the presence of CAFs was investigated using immunostaining in human thyroid cancer. Higher levels of α-SMA-positive areas were found preferentially localized at the tumor invasive front, in PTC and ATC. However, these studies did not find a specific or clear association between CAFs and a particular type of thyroid carcinoma [35]. Conversely, a very rare or absent infiltration of CAFs was described by α-SMA immunostaining in a study performed on patients with ATC [36]. Interestingly, a PTC murine model of oncogenic BRAFV600E activation in thyroid follicular cells showed increased fibroblast infiltration in thyroid cancer compared to normal thyroid tissue, in agreement with most observations in human tissue [37]. A higher density of α-SMA+ fibroblasts was also found in a mouse model of PTC where thyroid-specific expression of oncogenic BRAF is activated and Pten is lost (BRAFV600E/Pten−/−/TPO-Cre) [38]. Thus, these studies demonstrate recruitment and infiltration of CAFs into the TME in different types of thyroid carcinomas.
Once CAFs are accumulated in the TME, evidence suggests that they may subsequently continue to be attracted and/or activated in response to paracrine factors released from the neighboring tumor cells. For example, platelet-derived growth factor (PDGF), interleukin-6 (IL-6), reactive oxygen species (ROS), and transforming growth factor-β (TGF-β), among others, are key regulators of fibroblast recruitment and activation [8–11].
How are thyroid CAFs generated?
It has been established that interaction between cancer cells and fibroblasts can promote the CAF phenotype in diverse tumor types. Unfortunately, the biology of tumor–stroma crosstalk in human thyroid cancer is largely unexplored. In a study performed on patients with PTC, a significant correlation was found between higher TGF-β1 in cancer cells and increased α-SMA levels in surrounding fibroblasts [39]. To recapitulate the effects of tumor cell secretome on stromal cells, our laboratory utilized in vitro co-cultures of human thyroid cancer and human fibroblast cell lines. We found that thyroid cancer cell-derived conditioned media (CM) reprogramed the thyroid fibroblast phenotype (Fig. 1) [40]. As mentioned before, CAFs are more proliferative than quiescent fibroblasts. Indeed, the number of human fibroblasts was significantly increased by treatment with thyroid cancer cell-derived CM [40]. Reprogramming fibroblasts into CAFs also involves alterations in their phenotype. We also demonstrated that thyroid cancer cell-derived CM induced up-regulation of CAF markers such as PDGFR-β, α-SMA, and vimentin in human thyroid fibroblasts [40]. It has been demonstrated that, when transformed, CAFs show important metabolic alterations. In fact, glucose transporter 1 (GLUT-1) was markedly increased in human thyroid fibroblasts when cultured with thyroid cancer cell-derived CM (Fig. 1) [40]. Interestingly, we found that in addition to IL-6 and ROS, thyroid cancer cells secreted high amounts of PDGF into the media. All these soluble factors could serve as causative molecules that activate fibroblasts [40]. In line with our findings, Jolly et al. [38] reported that CM collected from thyroid cancer cells generated from BRAFV600E/Pten−/−/TPO-Cre mice significantly increased proliferation and migration of murine CAFs. Collectively, these data suggest that tumor-derived soluble factors can induce a CAF phenotype in thyroid carcinoma (Fig. 1). Nonetheless, many key questions still remain unsolved. For example, in addition to the paracrine signals generated by tumor cells, can cues released by other cells in the TME also contribute to fibroblast activation? Further studies are required to explore other TME-derived factors in thyroid cancer cells as well as the pathways involved in phenotypic reprogramming of human thyroid fibroblasts.
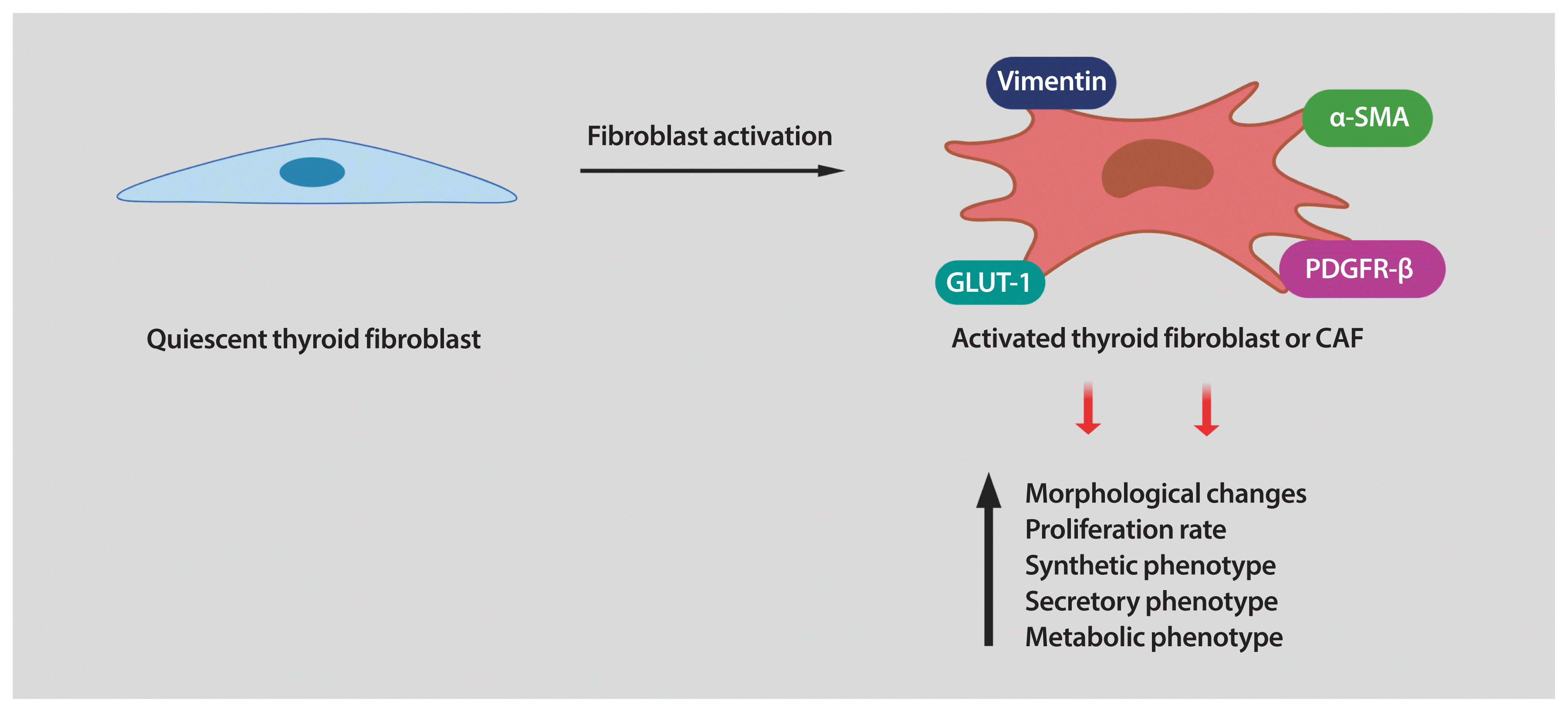 | Fig. 1Activation of thyroid fibroblasts. Cancer cells produce cytokines, chemokines, and growth factors to stimulate the transformation of quiescent or resting fibroblasts into activated cancer-associated fibroblasts (CAFs). Reprogramming of CAFs involves the increase in the levels of α-smooth muscle actin (α-SMA), vimentin, and platelet-derived growth factor receptor β (PDGFR-β), among other possible CAF markers. In addition, activated fibroblasts increase their proliferative properties and reprogram their secretory and metabolic phenotypes. Graphic created using BioRender. GLUT-1, glucose transporter 1.
|
Crosstalk between tumor cells and CAFs: tumor-stimulatory effects of CAFs in thyroid cancer
As tumors grow, CAF cell numbers increase and accumulate. Once CAFs are reprogrammed, they influence most of the hallmark capabilities of cancer cells, playing key roles in the process of cancer development and progression through a variety of mechanisms. CAFs can exert their tumorigenic functions directly by cell–cell contact and indirectly by utilizing secreted factors. The array of functions and the mechanisms by which CAFs promote tumor progression are outside the scope of this review, but have been discussed in detail elsewhere [8–11,32]. However, it is important to point it out that CAFs are a considerable source of multiple secreted factors. So, we will focus on the CAF secretome and how it influences cancer cell behavior to support malignant thyroid tumor growth.
What are the functions of CAFs in thyroid carcinogenesis?
Only a few studies have provided direct evidence of the pro-tumorigenic role of CAFs in thyroid cancer. Using an in vivo subcutaneous model, Saitoh et al. [41] demonstrated the crucial role of the interaction between thyroid tumor cells and skin fibroblasts to promote the growth of tumors, in rats. According to the model, co-injection of tumorigenic Fisher rat thyroid follicular cells (FRTL-Tc) mixed with skin fibroblasts resulted in subcutaneous tumors larger than those derived from FRTL-Tc cells alone. Interestingly, the authors reported that fibroblasts enhance the growth of thyroid carcinoma by secreting soluble factors, although they did not identify any of them [41]. Another important contribution in understanding the functional effects of soluble factors released by CAFs on thyroid tumor progression has recently been provided by our laboratory. By using in vitro co-cultures, we have found that thyroid cancer cells treated with CAFs CM showed increased proliferation and invasion. In addition, we determined that soluble factors secreted by CAFs were able to activate epithelial-to-mesenchymal transition (EMT) in thyroid cancer cells. Classical mitogens for epithelial cancer cells, such as IL-6, were highly expressed and secreted by CAFs. We also found that ROS are abundantly produced by pro-tumorigenic CAFs and secreted into the media [40]. Secretion of IL-6 and ROS by CAFs, among other soluble factors, are believed to promote thyroid tumor aggressiveness.
Another distinctive feature of CAFs is their ability to produce and deposit ECM. Alterations in the composition and cross-linking of the ECM change the stiffness of the tissue, thereby critically regulating tumorigenesis. The main constituents of the ECM are collagens [9]. Twenty-eight different collagens have been identified [42], and their assembly is highly regulated by the activity of enzymes, such as lysyl oxidase (LOX) [43]. Interestingly, LOX, an enzyme responsible for collagen cross-linking, was found to be highly expressed in aggressive thyroid carcinomas as compared to DTCs and normal thyroid tissues [35,44]. In addition, LOX inhibition significantly reduced the migration and invasion of ATC cell lines [44]. A recent study reported that BRAF-mutant tumors with high LOX expression were associated with more aggressive disease in thyroid cancer [45]. Consistent with these observations, in thyroid tumors from BRAFV600E murine models LOX expression levels were significantly higher than in normal tissue [38]. However, our current understanding of LOX and how it interacts with CAFs in thyroid cancer is still primitive, and more research is needed in order to design potential novel anti-tumor strategies based on LOX targeting for thyroid cancer.
Together, thyroid CAFs participate in a dynamic and interactive interplay with thyroid neoplastic cells. By secreting soluble factors (ROS, PDGF, and IL-6) thyroid tumor cells activate quiescent fibroblasts into CAFs. After activation, CAFs initiate the secretion of soluble factors and increase the ECM production, interacting with thyroid cells to foster malignant thyroid tumor growth by promoting cell proliferation, invasion, and EMT (Fig. 2).
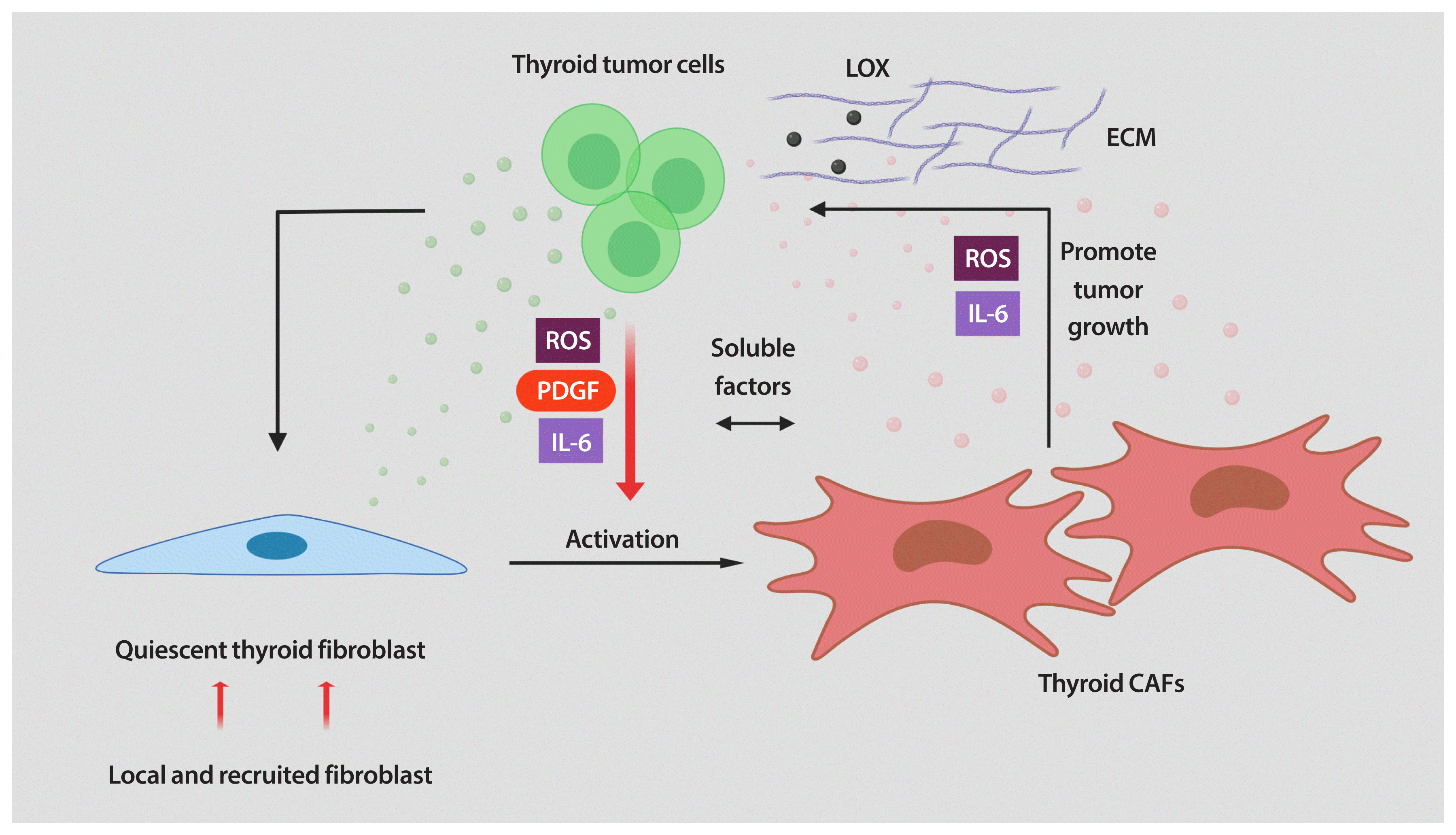 | Fig. 2A schematic model describing the dynamic interplay between cancer-associated fibroblasts (CAFs) and thyroid tumor cells. Thyroid tumor progression needs a positive feedback between CAFs and cancer cells. Cancer cells induce and maintain the fibroblasts’ activated phenotype which, in turn, produces a series of growth factors and cytokines that sustain thyroid cancer progression by promoting cell proliferation and cell invasion. Graphic created using BioRender. IL-6, interleukin-6; ROS, reactive oxygen species; PDGF, platelet-derived growth factor; LOX, lysyl oxidase; ECM, extracellular matrix.
|
Implications of targeting thyroid CAFs: therapeutic perspectives
Thyroid carcinoma, the most common form of endocrine malignancy, has an increasing incidence worldwide. While DTCs usually have a good prognosis, ATC is one of the most lethal solid tumors known. Conventional treatments of advanced thyroid cancer remain very challenging, and additional research is needed to find new treatment strategies to improve patient outcome.
Accumulating evidence indicates that CAFs exert pro-tumorigenic functions, making the different mechanisms that CAFs use to stimulate cancer cells exceptional therapeutic targets for cancer treatment [46]. Most strategies to manipulate CAFs to treat cancer focus on interfering with activation of CAFs or inhibiting CAF functions. Several combinatorial strategies targeting TGF-β, which regulates the activation of CAFs, are currently being tested in clinical trials. For example, galunisertib, a TGF-β receptor inhibitor, resulted in improved survival of patients with pancreatic cancer [47]. Inhibiting oncogenic signals secreted by CAFs by inhibitors or antibodies represents another attractive therapeutic strategy. Inhibitors of IL-6 are currently in use in different clinical trials and may be useful in combination therapies to treat cancer [48]. These observations highlight the potential of a wide range of therapeutic strategies targeting CAFs.
Go to :
CONCLUSIONS
The role of the TME, particularly CAFs, in the development of thyroid cancer has long been underestimated. There is now more evidence clearly suggesting that interactions between CAFs and cancer cells are involved in thyroid cancer progression. Indeed, some of the mechanisms that these cells of the TME use to promote thyroid tumor progression have recently been identified. These findings have made CAFs a promising new therapeutic target in thyroid cancer. Even so, numerous unanswered questions remain regarding CAFs in the thyroid cancer field. We believe that better understanding of thyroid CAFs’ activation and function, as well the relationship between CAFs and thyroid cancer cells, is needed to design improved anti-cancer therapies in the future. These novel strategies can then be combined to enhance thyroid cancer patient outcome.
Go to :
 XML Download
XML Download