

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Since the human telomerase reverse transcriptase (TERT) gene was first cloned in 1997 [1,2], extensive efforts have been made to reveal the mechanisms of TERT reactivation. With recent advances in high-throughput sequencing technologies, these mechanisms have been actively researched, with a particular focus on the genomic alterations involved in activating TERT in various cancers. Based on the recent pan-cancer analysis of The Cancer Genome Atlas (TCGA), the prevalence of TERT promoter mutations in cancers is reported to be 0% to 89%, with the highest rates in glioblastoma (89%), skin melanoma (72%), and bladder cancer (70%) [3]. In addition, TERT amplification is found in 0% to 22% of cancers, most frequently in ovarian cancer (22%) and adrenocortical carcinoma (15%). Among the subtypes of thyroid cancer, TERT promoter mutations are more common in poorly-differentiated thyroid cancer (PDTC; 21% to 47%) and anaplastic thyroid cancer (ATC; 55% to 73%) than in differentiated thyroid cancer (DTC), such as papillary thyroid cancer (PTC; 4% to 26%) and follicular thyroid cancer (FTC; 6% to 36%) [4,5]. Moreover, TERT promoter mutations often coexist with the BRAFV600E mutation, both in PTC (odds ratio [OR], 2.4) and in PDTC/ATC (OR, 2.4) [6]. Thus, efforts are ongoing to decipher the mechanisms of its association with other oncogenes and clinical outcomes. Reactivation of TERT and coexistence with BRAFV600E have been reported to be associated with poor prognosis in several cancers, particularly in thyroid cancer [5,7]. Therefore, understanding the mechanisms by which TERT affects tumor aggressiveness is important for improving the prognosis of these cancers. In this review, we present an overview of recent insights into the mechanisms of TERT reactivation, as well as its interactions with BRAF in human cancers, including thyroid cancer.
Go to : 
TERT REACTIVATION IN TUMORIGENESIS
Canonical functions of TERT: lengthening of telomeres
Key protection mechanisms against cancer are the repression of telomerase and the maintenance of short telomeres [8]. In most differentiated human cells, telomerase is silenced by transcriptional repression of the TERT gene, which encodes its catalytic component [9]. The lack of telomerase leads to progressive telomere erosion in dividing human cells. When telomeres have become critically short, they are detected by the DNA-damage repair machinery, and the cell dies or reaches a permanent growth arrest stage known as replicative senescence [10]. The senescence response is a potent tumor suppressive mechanism [11]. As infinite proliferation is a hallmark of malignant cells, up to 90% of human cancers overcome the senescence barrier by reactivating telomerase [8,12]. The telomere length is maintained by the reactivation of telomerase/TERT in most tumor cells, while a small proportion acquire immortality through the telomerase-independent alternative lengthening of telomeres (ALT) mechanism, a homologous recombination-based process (Fig. 1) [12,13].
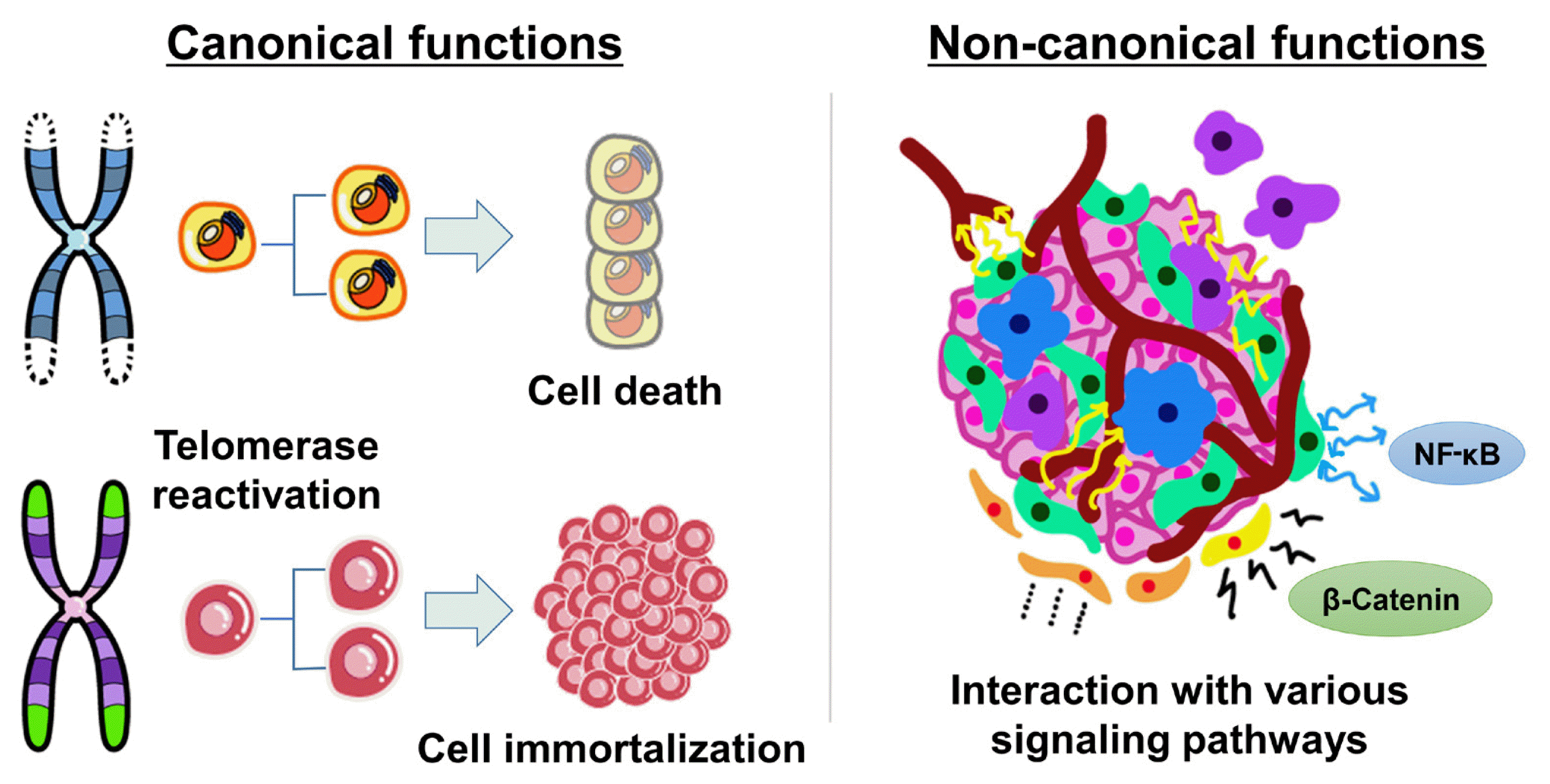
Non-canonical functions of TERT: interactions with oncogenic signaling pathways
TERT has also been shown to exhibit multiple biological activities, independently of its telomere lengthening function, regulating various genes and signal pathways involved in the hallmarks of cancer, such as cell proliferation, angiogenesis, resistance to apoptosis, inflammation, invasion, and metastasis (Fig. 1) [14, 15]. Intriguingly, nuclear factor-κB (NF-κB) and β-catenin, master regulators of many oncogenic targets directly or indirectly driving various hallmarks of cancer, are known activators of TERT expression, and TERT regulates their transcriptional activities, forming a feed-forward loop in cancer cells [16]. TERT interacts with NF-κB p65, activating NF-κB target genes, including interleukin (IL)-6, tumor necrosis factor alpha, and IL-8, cytokines known to sustain inflammation and cancer progression, and upregulating matrix metalloproteinases, which are important for metastasis [16,17]. In addition, TERT directly interacts with the Wnt/β-catenin signaling pathway and amplifies its transcriptional output, stimulating the epithelial-mesenchymal transformation and stemness of cancer cells, which contribute to invasion and metastasis [18,19]. Therefore, TERT reactivation contributes to cancer development and progression by both telomere lengthening-dependent and independent mechanisms.
Go to :
MECHANISMS OF TERT REACTIVATION IN CANCER
TERT reactivation can be caused by mechanisms including genetic (promoter mutations, amplifications, and rearrangements) and epigenetic (DNA methylation, histone acetylation/deacetylation, and non-coding RNAs) events (Fig. 2). Advances in high-throughput next-generation sequencing technologies have enabled researchers to unravel numerous genomic aberrations in various human malignancies. In an integrated analysis of 31 cancer types derived from the TCGA cohort, 95% of TERT-expressing samples had TERT aberrations, including TERT promoter mutations (31%), TERT amplification (3%), TERT structural variants (3%), TERT promoter structural variants (5%), or TERT promoter methylation (53%) [3].
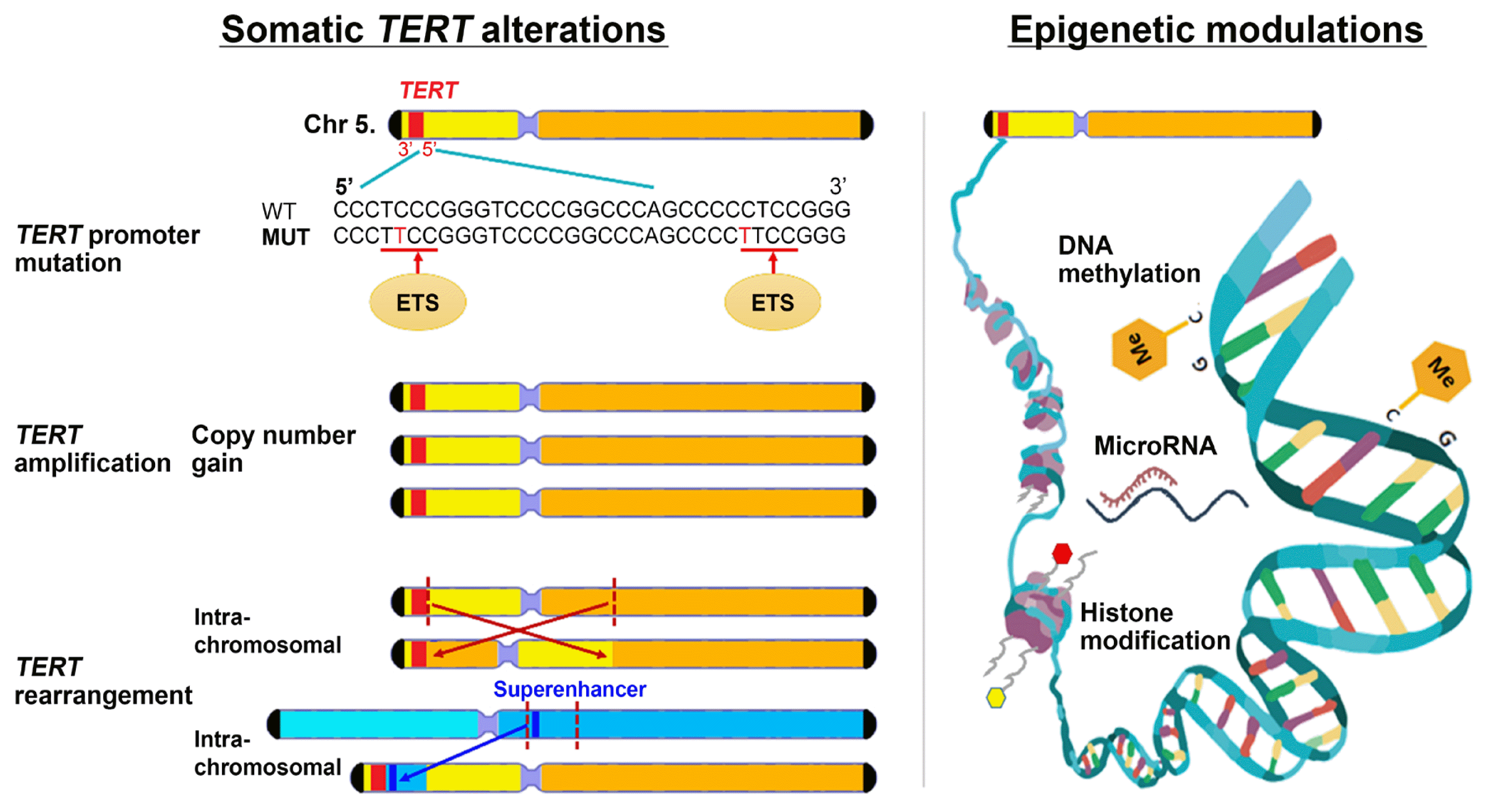 | Fig. 2Mechanisms of telomerase reverse transcriptase (TERT) reactivation in cancer. TERT reactivation can occur by somatic TERT alterations (promoter mutation, amplification, and rearrangement) and epigenetic modulation (DNA methylation, non-coding RNA, and histone modification). WT, wild-type; MUT, mutant; ETS, E-twenty six.
|
TERT promoter mutations in human cancers
TERT promoter mutations are the most common non-coding mutations in human cancer. Since 2013, when two studies on melanoma were published [20,21], research exploring TERT promoter mutations in various cancers, including thyroid cancer, has robustly increased [22]. These mutations are recurrent C>T transitions occurring at chr5:1295228 (−124 or C228T) or chr5:1295250 (−146 or C250T) within the core promoter of TERT. TERT promoter mutations upregulate TERT transcription by creating a de novo binding site for E-twenty six (ETS) transcription factors [20,23] and recruiting these factors to the TERT promoter in a mutation-dependent manner [23,24].
The mutations can arise as in the context of malignant transformation (e.g., in the liver) [25], but overall, they represent a late event in most cancers. Tumor cells harboring TERT promoter mutations have short telomeres, suggesting that these mutations are a late event of tumorigenesis, after telomeres have become critically short [3,26,27]. Considering that the selective pressure for TERT promoter mutations should be strongest near replicative senescence after multiple cycles of replication, it is logical that these mutations are acquired late. In cancers caused by environmental exposure (e.g., skin and bladder), TERT promoter mutations can represent an early event [28]. Such alterations are not found in some cancer types, such as gastrointestinal or hematologic malignancies, which arise from tissues with high rates of self-renewal [3]. Because these tissues have intrinsic telomerase activity, allowing them to maintain telomere length over many cell divisions, selection for TERT promoter mutations may be less important for them.
In a pan-cancer study including 31 cancer types, TERT promoter mutations were detected in 27% (range, 0% to 89%) of the analyzed cases [3]. TERT promoter mutations have been demonstrated across a range of malignancies, namely in the central nervous system (89% of glioblastomas and 45% of lower-grade gliomas), skin (72% of melanomas), urothelial bladder (70%), liver (45%), head and neck (24%), and thyroid (10% of PTCs) [7]. The prognostic impacts of TERT promoter mutations and their potential use as clinical biomarkers in various cancers have been evaluated. TERT promoter mutations are associated with decreased overall survival in bladder cancer [29], brain tumors [30,31], melanoma [32], laryngeal cancer [33], as well as in thyroid cancer [5,34–37], with hazard ratios of 1.3 to 21.1.
TERT amplification in human cancers
The amplification of oncogenes is a frequent event in cancer and typically causes gene overexpression. Since TERT amplification in human cancer was first identified in 2000 [38,39], many studies have shown this genomic event in many different types of malignancies, and in recent years, copy number variations have been more successfully detected by next-generation sequencing. In the TCGA study, TERT focal amplifications were found in 4% (range, 0% to 22%) of all examined tumors, with high frequencies in ovarian cancer (22%), adrenocortical carcinoma (15%), esophageal cancer (14%), and non-small cell lung cancer (13%) [3]. TERT amplifications play an important role in the diagnosis of various solid tumors, including breast phyllodes tumors, non-small cell lung cancer, and bladder cancer. TERT amplifications also predict a poor prognosis in breast cancer [40,41], bladder cancer [42], non-small cell lung cancer [43], and acral-lentiginous melanoma [44].
TERT rearrangements in human cancers
TERT rearrangements involving the gene promoter or the gene body are another TERT reactivation mechanism that occurs in human cancers. Of particular note, in the majority of TERT promoter rearrangements, super-enhancers directly overlap with the juxtaposed TERT coding sequence. This repositioning of enhancer elements through rearrangement leads to massive transcriptional upregulation of TERT and strong chromatin remodeling of the affected region [45]. In support of this hypothesis, TERT promoter rearrangements were reported to result in the highest TERT expression level among somatic TERT aberrations [3,46]. TERT rearrangement with telomerase activation was first reported in an immortal fibroblast cell line in 2009 [47]. Recently, using high-throughput sequencing, structural variations involving rearrangements of the TERT gene have been identified in many cancer types, including neuroblastoma [45,48,49], hepatocellular carcinoma [50], kidney chromophobe cancer [51], sarcoma, and prostate cancer [3]. TERT rearrangements have been most comprehensively analyzed in neuroblastoma, and recurrent rearrangements proximal to the TERT gene have been reported in high-risk tumors with a poorer outcome [48,52].
Epigenetic mechanisms regulating TERT in human cancers
Epigenetic mechanisms, which consist of alterations other than direct DNA sequence changes, can modulate TERT transcription. TERT gene transcription is regulated not only by the assembly of transcription factors at promoter or enhancer regions, but also the modification of their accessibility to DNA, a process controlled by epigenetic mechanisms including DNA methylation, histone methylation, and histone acetylation [53]. The TERT promoter region is rich with binding motifs for multiple transcript factors including MYC proto-oncogene, specificity protein 1 (SP1), ETS, activator protein 1 (AP1), signal transducer and activator of transcription 3 (STAT3), and tumor protein p53 [54,55], and also contains binding sites for repressors such as CCCTC-binding factor (CTCF), SIN3 transcription regulator family member A (SIN3A), and MYC-associated zinc finger protein (MAZ) [56,57]. In general, hypermethylation of CpG islands of gene promoters is associated with gene silencing, while hypomethylation of CpG islands is associated with gene overexpression. However, TERT promoter methylation has been reported to be paradoxically correlated with TERT overexpression in most TERT-positive tumor cells, while the absence of TERT methylation has been found in some TERT-negative tumors and TERT-negative normal cells [58]. An explanation for this is that the unmethylated promoter sequence favors repressor binding; therefore, the hypermethylated state interferes with the binding of transcriptional repressors, resulting in upregulated TERT expression and telomerase activity [59]. In addition, associations between TERT promoter hypermethylation and poor prognoses have been reported in brain tumors [60], pancreatic [61] and prostate cancers [62], and paragangliomas [63]. Other epigenetic mechanisms including histone modifications [64–66] and non-coding RNA interactions with TERT [58] may also be involved in TERT regulation.
Go to :
MECHANISMS OF TERT REACTIVATION IN THYROID CANCER
TERT promoter mutations in thyroid cancer
An association between TERT promoter mutation and increased TERT expression has been demonstrated in thyroid cancer [3,67,68]. The mechanism through which TERT expression is enhanced by TERT promoter mutations in thyroid cancer is that ETS transcription factors selectively bind and activate the mutant TERT promoter [68,69], which is consistent with the mechanisms proven in other cancers. The prevalence of TERT promoter mutations is significantly higher in ATC and PDTC than in DTC [4,5]. Moreover, the TERT expression levels induced by promoter mutations have also been reported to be higher in ATC than in DTC, which may be due to the expansion of subclones with TERT promoter mutations in ATC [46,70]. In the pan-cancer study of the TCGA cohort, the proportion of samples without TERT expression was higher in PTC than in cancers of other organs; a total of 22% of analyzed tumors had neither detectable TERT expression nor ALT-related abnormalities, and 79% of PTC cases did not have them [3]. TERT promoter mutations are well-known to be associated with high-risk clinicopathologic characteristics and poor outcomes of DTC [5,34–37]. TERT expression has also been reported to be correlated with aggressive tumor behavior and a poorer prognosis in DTC, even independent of TERT promoter mutation status [67,71–73].
TERT amplification in thyroid cancer
Few studies have investigated TERT aberrations other than TERT promoter mutations in thyroid cancer. In a recent study of follicular thyroid tumors, TERT copy number gain was observed in six of 77 (8%) FTCs, four of 19 (21%) follicular tumors of uncertain malignant potential (FT-UMPs), and two of 43 (5%) follicular adenomas (FAs), and these proportions were not significantly different between groups [71]. Moreover, TERT copy number gain was associated with an increased risk for recurrence in FTC after adjusting for covariates. Another recent study investigated mechanisms of TERT activation in various types of thyroid cancer including PTC, FTC, Hürthle cell cancer (HCC), medullary thyroid cancer (MTC), and PDTC/ATCs [74]. Increased TERT copy numbers were found in one of 107 (0.9%) PTCs, two of 22 (9.1%) FTCs, four of 29 (13.8%) HCCs, 0 of 22 (0%) MTCs, and two of four (50.0%) PDTC/ATCs. Thus, the prevalence of TERT amplification was higher in aggressive cancer types, such as HCC and PDTC/ATC, although the sample size was limited. Interestingly, among thyroid cancer cases with TERT copy number gain, co-occurrence with TERT promoter mutations (C228T) was found in two of four HCC and one of two PDTC/ATC samples.
TERT rearrangements in thyroid cancer
TERT fusion genes were reported in one PTC case in the TCGA study (myotubularin-related protein 12 [MTMR12]-TERT) and in one widely-invasive FTC (wiFTC) case of our previous study (phosphodiesterase 8B [PDE8B]-TERT), which were detected using RNA-sequencing [46]. These intra-chromosomal rearrangements showed the typical characteristic of fusion genes that TERT expression after the breakpoint was markedly increased. Moreover, an inter-chromosomal translocation at an upstream region of TERT (t[2;5][2q:5p]) was demonstrated in a wiFTC case, using RNA-sequencing and whole-genome sequencing. This rearrangement remarkably enhanced TERT expression by super-enhancer hijacking. Like other types of cancer, TERT expression was notably upregulated in thyroid cancer samples with TERT structural rearrangements in comparison to those with promoter mutations [46].
Epigenetic mechanisms regulating TERT in thyroid cancer
Epigenetic mechanisms can regulate TERT transcriptional activity in thyroid cancer. Lee et al. [59] identified the 52 CpG-containing TERT hypermethylated oncological region (THOR) as a cancer-associated epigenetic mechanism of TERT upregulation in various human tumor types and found frequent (>45%) cancer-associated DNA hypermethylation in most cancer types. However, exceptionally, in thyroid tumors (including 31 PTCs, two FTCs, and five FAs), only one case of PTC (3%) showed high THOR methylation, while the other 97% of thyroid tumors had hypomethylated THOR. In the aforementioned study of follicular thyroid tumors, TERT promoter hypermethylation (methylation index >18%) was found in 25 of 77 (32%) FTCs, three of 25 (12%) FT-UMPs, and 0 of 42 (0%) FAs, and was associated with recurrence of FTC after adjusting for covariates [71].
Go to :
MECHANISMS OF SYNERGISTIC INTERACTION BETWEEN BRAFV600E AND TERT PROMOTER MUTATIONS IN CANCER PROGRESSION
TERT promoter mutations in thyroid cancer have been shown to be associated with the BRAFV600E mutation, and the coexistence of these two mutations exerts a synergistic negative effect on clinical outcomes [5,35–37,75]. The co-occurrence of BRAFV600E and TERT promoter mutations has been identified in thyroid cancer, melanoma, and glioma, and appears to be associated with tumor aggressiveness [76,77]. Therefore, the mechanisms underlying the interaction between these mutations have been explored (Table 1).
Table 1
Studies of Mechanisms Underlying the Synergistic Interactions between BRAFV600E and TERT Promoter Mutations in Cancer
| Study | Study subjects (n) | Suggested mechanism |
|---|---|---|
| Melanoma | ||
| Vallarelli et al. (2016) [78] | Cell lines of melanoma (8) and melanocytes (1) | TERT promoter mutations form a direct link between TERT expression and MAPK pathway activation due to BRAF or NRAS mutations via the transcription factor ETS1 in melanoma. |
| Li et al. (2016) [79] | Cell lines of melanoma (7) | RAS-ERK signaling activation by BRAFV600E or NRAS mutations maintains an active chromatin state at the mutant TERT promoters in melanoma, facilitating the recruitment of RNA polymerase II and thereby leading transcriptional activation of TERT. |
|
|
||
| Glioma | ||
| Gabler et al. (2019) [80] | Cell lines of glioma (12) | BRAFV600E-induced expression and phosphorylation of ETS1 enhance TERT expression and TERT promoter activity in gliomas with both mutations of BRAFV600E and TERT promoter. |
|
|
||
| Thyroid cancer | ||
| Liu et al. (2018) [69] | Cell lines of thyroid cancer (8), melanoma (7), colon cancer (1), embryonic kidney (1), thyrocytes (1) | BRAFV600E/MAPK pathway promotes phosphorylation and binding of the FOS transcription factor to the GABP promoter, increasing GABPB expression and formation of the GABPA-GABPB complex. This complex selectively binds and activates the mutant TERT promoter, upregulating TERT expression in human cancer. |
| Song et al. (2019) [68] | Tissue samples of papillary thyroid cancer (331; 266 from TCGA and 65 from their own cohort); cell lines of thyroid cancer (8; 2 papillary and 6 anaplastic thyroid cancers) and thyrocytes (2) | ETS transcription factors, such as ETV1, ETV4, and ETV5, which are upregulated by BRAFV600E/ MAPK pathway activation, selectively bind to the mutant TERT promoter in thyroid cancer. TERT expression is increased by the coexistence of BRAFV600E and TERT promoter mutations, and the pathways related to immune response or adhesion molecules are activated by TERT expression. |
| Bullock et al. (2019) [81] | Tissue samples of normal thyroid (59) and papillary thyroid cancer (498; all from the TCGA cohort); cell lines of thyroid cancer (3; 1 papillary and 2 anaplastic thyroid cancers) and thyrocytes (1) | ETV5 is the most transcriptionally upregulated ETS gene in papillary thyroid cancer and is strongly correlated with BRAF and RAS mutational status. ETV5 preferentially binds the mutant TERT promoter allele and enhances TERT transcription, cooperating with FOXE1 to further increase TERT promoter activity. |
![]()
In a melanoma study, the ETS1 transcription factor, upregulated as a downstream target of the activated mitogen-activated protein kinase (MAPK) pathway due to BRAF or NRAS mutations, increased TERT transcription in melanoma cells carrying TERT promoter mutations [78]. Moreover, another study of melanoma showed that aberrant activation of RAS-extracellular signal-regulated kinase (ERK) signaling by BRAFV600E or NRAS mutations maintained an active chromatin state and facilitated the recruitment of RNA polymerase II, leading to transcriptional activation of TERT at the mutant TERT promoter [79]. A recent study of glioma showed that, in BRAFV600E/TERT promoter double-mutant gliomas, the expression and phosphorylation of ETS1 selectively binding to the mutant TERT promoter was downregulated by BRAF inhibition, and TERT expression was reduced by an ETS factor inhibitor [80]. Thus, this study suggested that the ETS transcription factor may be a promising therapeutic target in double-mutant gliomas.
Mechanisms of interaction between BRAFV600E and TERT promoter mutations in thyroid cancer
Liu et al. [69] investigated the molecular mechanisms underlying the interactions between BRAFV600E and TERT promoter mutations using cancer cell lines, primarily including thyroid cancer and melanoma cells. They reported that BRAFV600E activated the mutant TERT promoter by fos proto-oncogene (FOS), which is a downstream effector of MAPK signaling, and the GA-binding protein (GABP) complex. The BRAFV600E/MAPK pathway promoted phosphorylation and binding of the FOS transcription factor to the GABPB promoter, increasing GABPB expression and formation of the GABPA-GABPB complex. This complex selectively bound to the mutated TERT promoter and strongly upregulated TERT expression.
In our recent study of PTC, we confirmed that TERT mRNA expression was increased by the coexistence of BRAFV600E and TERT promoter mutations using RNA sequencing data from a relatively large number of PTC tumor samples (266 PTCs from TCGA and 65 PTCs from Seoul National University Hospital) [68]. The changes in the intracellular signaling pathways by BRAFV600E were further augmented by adding the TERT promoter mutation, and the pathways related to inflammation or adhesion molecules were upregulated by TERT expression, which is consistent with one of the non-canonical roles of TERT—namely, activating the NF-κB signaling pathway. Moreover, among the transcription factors that can regulate TERT promoter activity, the expression of ETS, especially ETS variant transcription factor 1 (ETV1), ETV4, and ETV5, showed the most significant difference in BRAFV600E-mutant PTCs. The mechanism was validated using thyroid cancer cell lines, as ETV1, ETV4, and ETV5 were upregulated by activation of the BRAFV600E/MAPK pathway and selectively bound to the mutant TERT promoter.
Bullock et al. [81] also identified that ETV5 was significantly upregulated in PTCs, especially in those harboring BRAFV600E or RAS mutations, and preferentially bound to the mutant TERT promoter with allele-specific affinity, although they investigated only a subset of ETS genes among all ETS transcription factors and did not check ETV1 and ETV4. Two subsequent studies [68,81] did not find an association between BRAFV600E and expression of GABPA, GABPB1, or GABPB2 in PTC samples. Moreover, a recent study found that GABPA was negatively associated with TERT expression and promoter mutations, and acted as a tumor suppressor in thyroid cancer [82]. This discrepancy between in vitro cell lines and tumors from PTC patients suggests a more complicated relationship of TERT promoter mutations with the ETS transcription factor family, which requires further investigation. However, it is at least recognized that the mechanism of synergistic effects between TERT promoter and BRAFV600E mutations on the aggressiveness of thyroid cancer may be explained by the activation of TERT transcription and telomerase activity, which results from binding of BRAFV600E-induced ETS transcription factors to the mutant TERT promoter.
Go to :
CONCLUSIONS
Since telomerase repression is a powerful mechanism of tumor suppression, TERT reactivation is found in most human cancers. TERT reactivation occurs by somatic TERT alterations and epigenetic modulations. Moreover, the recruitment, accessibility, and binding of transcription factors as activators or repressors also affect the regulation of TERT expression (Fig. 3).
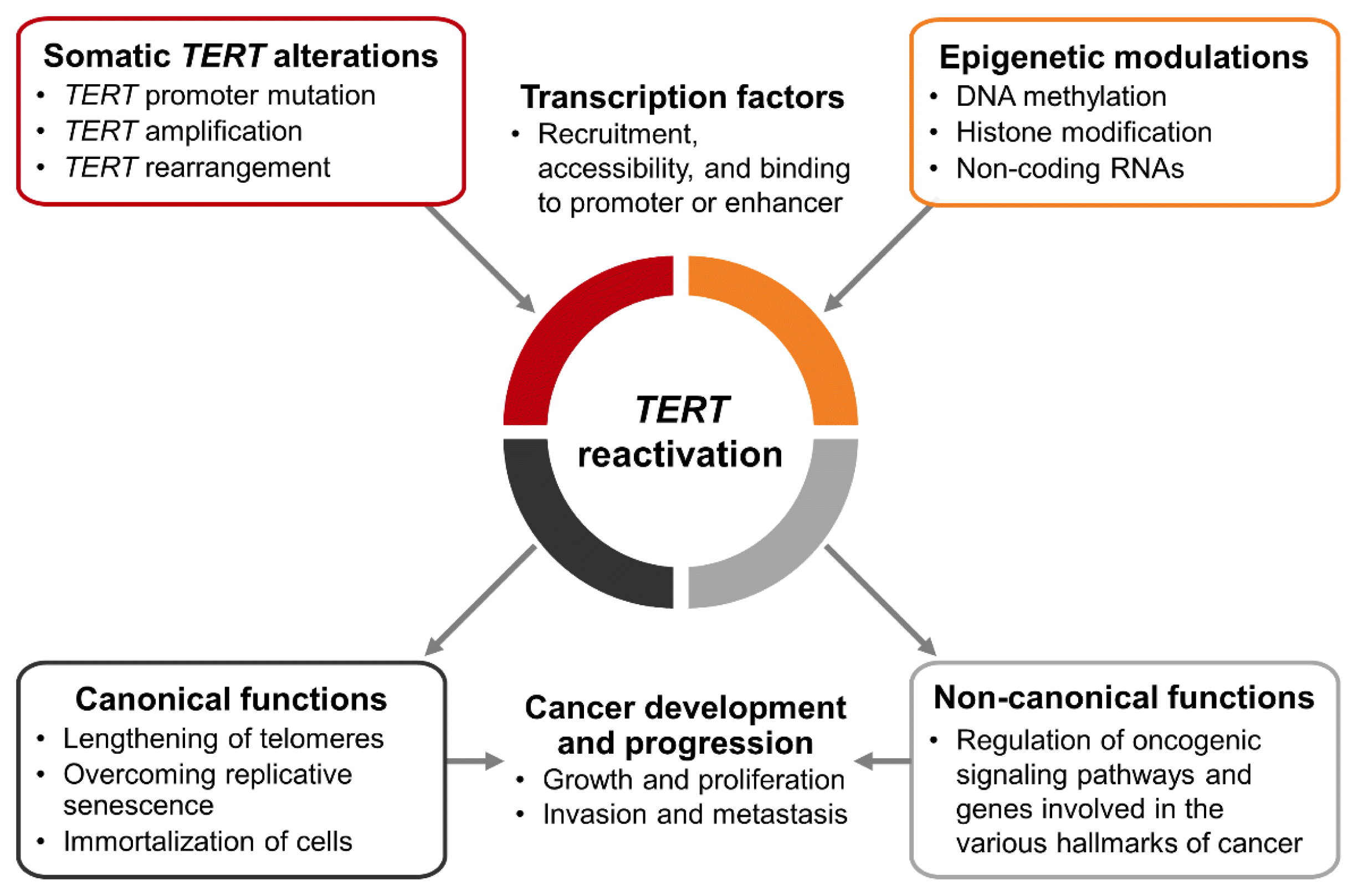
TERT promoter mutations are the most frequent non-coding alterations in human cancer, and have been detected in various types of cancer, including thyroid cancer. Other TERT aberrations (TERT amplification, rearrangement, and TERT promoter methylation) involved in telomerase activation are relatively less studied than TERT promoter mutations in thyroid cancer, and further research is therefore needed.
TERT reactivation causes cancer development and progression through telomere lengthening-dependent and independent ways, and has been reported to be associated with poor prognoses in various cancers. In melanoma, glioma, and thyroid cancer, in which the coexistence of BRAFV600E and TERT promoter mutations has been documented and investigated, these two mutations show synergistic interactions that contribute to poor outcomes. As the mechanism underlying this synergism, it has been demonstrated that activation of the MAPK signaling pathway by the BRAFV600E mutation creates an active chromatic state or enhances binding of ETS transcription factors at the mutant TERT promoter, thereby increasing TERT expression and telomerase activity. Understanding the mechanisms of TERT reactivation is important, as these insights could be used to prevent and treat refractory cancers with increased TERT expression.
Go to :
 XML Download
XML Download