

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
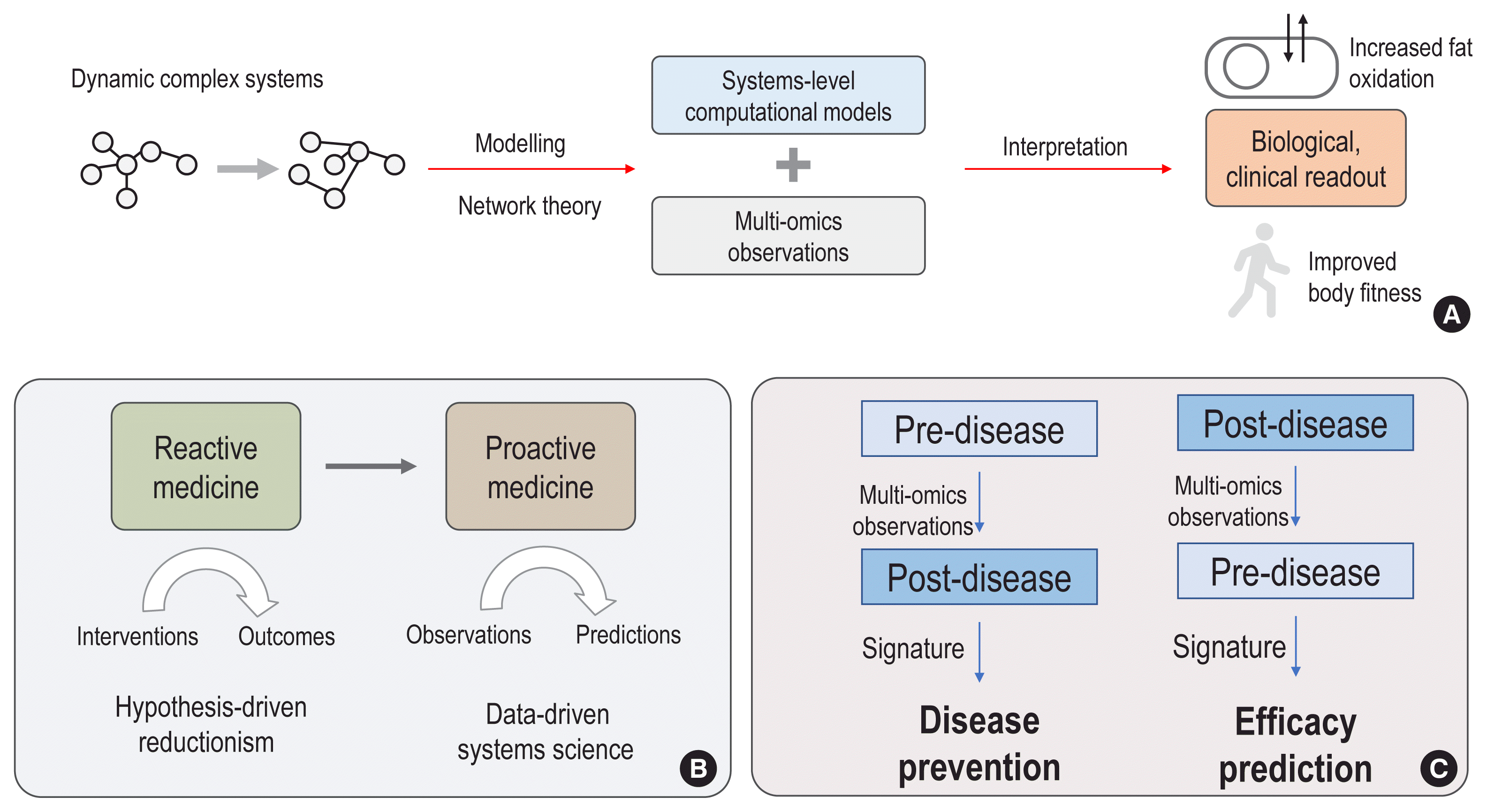
The human body maintains homeostasis through highly complex interactions of biochemical reactions, physiological processes, and environmental interactions at different scales [1]. These complex and dynamic properties have been overlooked in many disciplines of biology and medicine due to the lack of quantitative and systems approaches. Recently, many interesting strategies have been devised to simplify complex and dynamic biological problems into more understandable formulas (Fig. 1A) [2]. For example, network theory has been applied to cellular regulatory networks, uncovering key driver genes or modules of disease pathogenesis [3]. Genome-scale metabolic modelling (GEM) has been employed to explain metabolic phenotypes in various living systems, including human tissues and organs, the human gut microbiome, and bacteria engineered to produce biofuels and chemicals [4–12]. In addition, computational models and networks emulating biological systems have been established and freely shared by web-based services, including Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, Human Metabolic Atlas, and Tissue and Cancer Specific Biological Network (TCSBN) database [13–16]. These collaborative efforts will expand our understanding of medicine, enabling proactive medicine based on personalized data clouds—that is, systems medicine (Fig. 1B).
In recent years, with the boom of high-throughput omics data, human physiology has been extensively investigated through data-driven approaches. Based on longitudinal multi-omics profiling, we can identify individual signatures of pre- and post-disease states (Fig. 1C). For example, 108 healthy individuals were monitored for a year with personal multi-omics data, including metabolomics, proteomics, and gut metagenomics [17]. Based on multi-omics observations, subjects progressing from pre-disease states, such as elevated glucose or haemoglobin A1c levels, inflammation, and iron deficiency, were coached to change their lifestyle and diet to prevent poor health outcomes. Moreover, some multi-omics studies have identified individual signatures of the transition from post-disease to pre-disease states as a result of interventions. For example, the effects of an isocaloric low-carbohydrate diet in subjects with hepatic steatosis were investigated with multi-omics data, including metabolome, biopsy (liver) transcriptome, gut metagenome, and inflammatory markers [18]. Within 2 weeks after starting the low-carbohydrate diet, significant reductions of liver fat, plasma triglyceride levels, and inflammatory markers were observed despite no significant weight changes. In particular, genome-scale metabolic models (GEMs) have been provided as scaffolds to explain metabolic phenotypes by integrating metabolomics and transcriptomics data. A liver GEM predicted increased fluxes through fatty acid beta-oxidation and one carbon metabolism, which generates antioxidants and cofactors, including NAD+ and glutathione, for beta-oxidation, and these increased fluxes were also linked to increased folate production of the gut microbiome.
Such advances in systems medicine would eventually allow N-of-1 trials—that is, clinical trials designed for a single patient—based on the prediction of treatment efficacy and side effects (personalized and predictive medicine). Moreover, through systems biology tools and models, possible risk factors will be prevented through active participation of customers (preventive and participatory medicine). Personalized, predictive, preventive, and participatory medicine (P4 medicine) would thereby transform the current form of healthcare services in the near future. Recently, international consortia of systems medicine, including the European Association of Systems Medicine (EASyM) and the Coordinating Action Systems Medicine (CASyM), have started discussions on a roadmap for implementing systems approaches into medical practice [19,20], and also for systems medicine courses comprising multidisciplinary education and specific analytic skills [21].
In the following sections, we detail how systems biology methodology could help understand health and disease with concrete examples. Specifically, we explain (1) how GEM could identify tissue-specific or disease-specific metabolic signatures and new therapeutic compounds for personalized treatments; (2) how multi-omics data can be integrated into genome-scale models to identify biomarkers of dysregulated metabolism; and (3) how associations of the gut microbiome with diseases can be identified with quantitative and systems approaches.
COMPUTATIONAL MAPS OF HUMAN METABOLISM: GENOME-SCALE METABOLIC MODELS
All living organisms, including humans, survive by converting nutrients into energy and chemical substances used as the building blocks of cells [22,23]. All biochemical reactions that produce metabolites, energy, and redox power are strictly regulated in cells, especially by metabolic enzymes, and these reactions are interconnected, as different biochemical reactions share substrates. Because of the highly interconnected nature of metabolism, any changes in biological processes could affect metabolism, and therefore dysregulated processes in diseases could leave metabolic footprints [22].
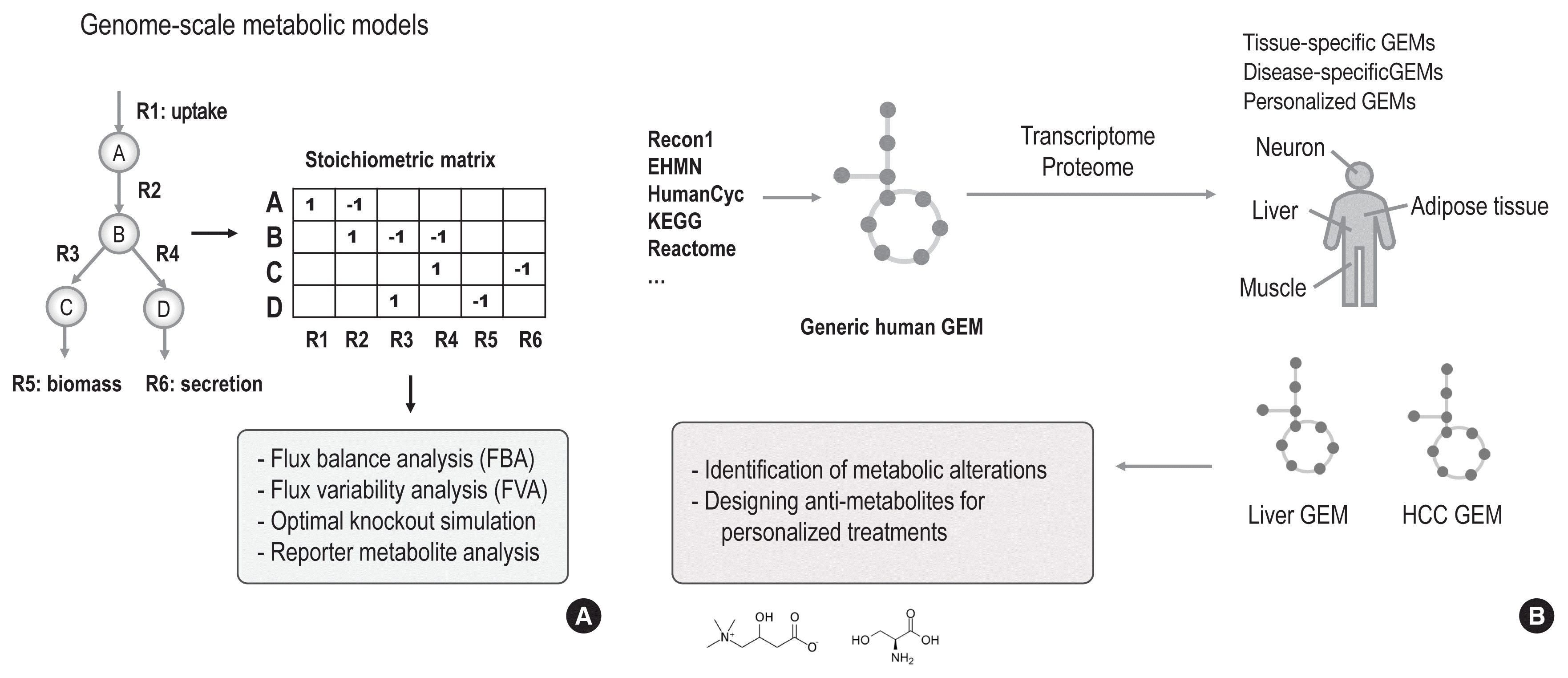
Recently, the systems biology community has established a computational “map” to explore the complexity of metabolism through GEMs (Fig. 2A). In these maps, all metabolites are connected through roads (“reactions”) and the traffic along all the roads are controlled by enzymes. Based on a metabolism map, we can estimate the possible degree of traffic (“flux”) by linear programming (or called linear optimization), through a process known as flux balance analysis (FBA). For example, bacteria regulate metabolic fluxes to maximize the production of biomass, so we can identify higher fluxes for all the roads (“reactions”) that contribute to the production of biomass by using FBA as an optimization method. However, in humans, metabolic demands vary across cells and tissues. Thus, an alternative strategy is to estimate fluxes by integrating information regarding enzymes, which function as “traffic controllers” [4,24, 25]. For example, when gene expression levels of glycolytic enzymes are higher in cancers, we could predict relatively higher fluxes of the corresponding enzymes.
Using this strategy, we can also generate tissue- and disease-specific metabolic “maps,” or GEMs composed of fluxes that may be maintained in given conditions (Fig. 2B) [4,25]. For example, hepatocyte, adipocyte, and myocyte GEMs were generated and analysed in the context of liver disease, obesity, and diabetes [26–28]. Using a hepatocyte GEM, serine deficiency was identified in non-alcoholic fatty liver disease (NAFLD) and elevated levels of branched amino acids in obese patients were identified using an adipocyte GEM [26,27,29].
Moreover, generating personalized GEMs could guide the treatment of individual tumors [25,30,31]. For example, dysregulated metabolites identified from personalized GEMs can be treated with anti-metabolites, which are analogue compounds of given metabolites that function as competitive inhibitors [25]. In hepatocellular carcinoma, 101 anti-metabolites have been predicted to suppress tumour growth in individuals, among which the anti-metabolite of L-carnitine, perhexiline, has had its inhibitory effect validated using the HepG2 cell line [25]. Furthermore, personalized GEMs for NAFLD have guided interventions to supplement metabolic co-factors (serine, N-acetyl-cysteine, nicotinamide riboside, and L-carnitine) to reduce liver fat [30,31], and its acute effects were observed based on plasma metabolomics and levels of inflammatory markers [32].
Lastly, the functional role of the human gut microbiome can be studied with GEMs. The human gut microbiome can significantly contribute to host metabolism by breaking down resistant fibres to generate short-chain fatty acids (SCFAs), such as acetate, propionate, and butyrate, and synthesizing essential amino acids and vitamins [33]. Its contribution to host metabolism can be studied with tissue-specific GEMs. For example, 28 tissue-specific GEMs from a mouse model were generated using proteomics data and the contribution of the microbiome to host metabolism was investigated. In short, by comparing gene expression levels between conventionally raised and germ-free mice, tissue GEMs predicted that host amino acid and glutathione metabolism would be different in conventionally raised mice, compared to germ-free mice [6,7]. Moreover, diet-induced metabolic changes caused by the microbiome can be studied with the state-of-the art algorithms of microbial community simulations using microbial GEMs, known as the Community And Systems-level INteractive Optimization (CASINO) toolbox [7]. Based on this microbial community algorithm, alterations of faecal and serum amino acid and SCFA levels were predicted and validated with faecal and blood metabolomics. In addition, personalized nutrition can be recommended based on the gut microbiome composition and diversity measure.
MULTI-OMICS INTEGRATION APPROACHES
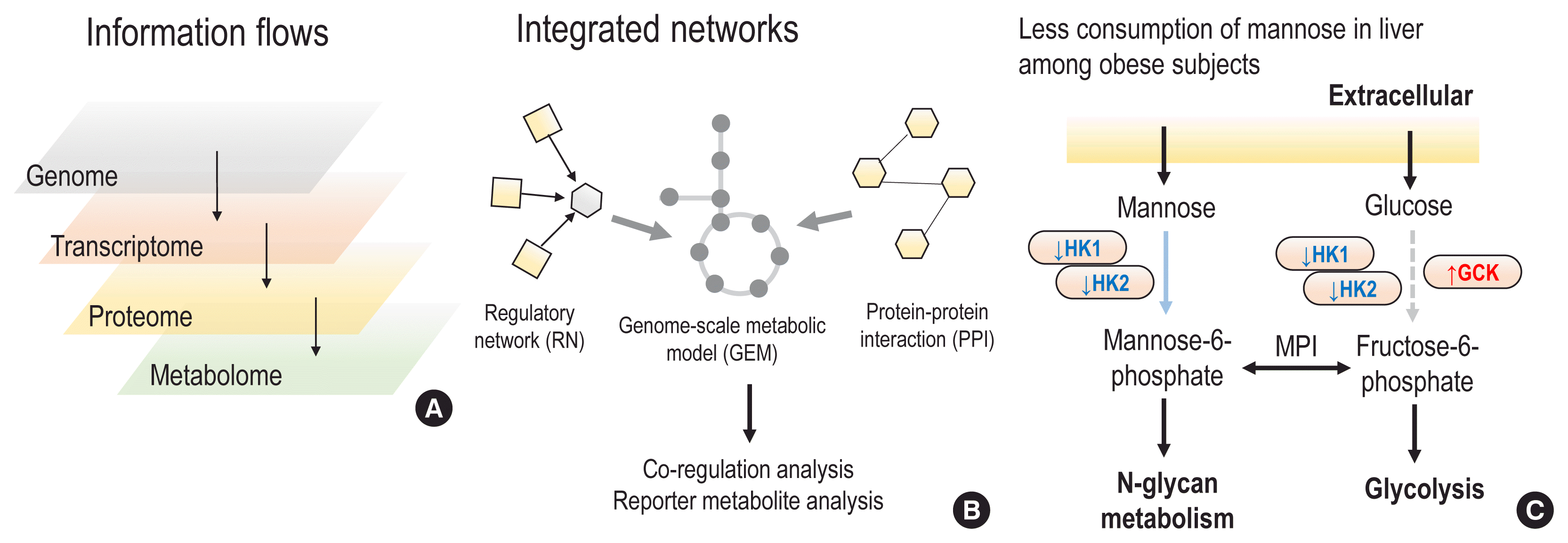
Following the information flow in the central dogma of molecular biology, any genomic, transcriptomic, and proteomic changes can be read out in the form of metabolomic changes (Fig. 3). Therefore, multi-omics observations would yield clear insights regarding which levels of omics initiate the downstream changes to metabolic alterations in chronic diseases. Currently, thousands of risk loci of diseases have been identified by genome-wide association studies (GWAS), but many of their linkages to pathophysiology have remained obscure. Recent studies have attempted to integrate GWAS findings with those of other omics studies, including RNA-seq, and by doing so have gained better insights into the identification of causal variations [34]. Likewise, multi-omics observations could identify how genetic, epigenetic, or transcriptional changes lead to metabolic alterations in complex diseases in a comprehensive manner.
Importantly, GEMs could serve as a scaffold for integrating biological networks and multi-omics datasets, thereby allowing us to trace the alterations of biomolecule abundance levels and their interactions in different omics layers. For example, by integrating tissue-specific biological networks into GEMs, such as hepatocyte, adipocyte, and myocyte GEMs, tissue-specific metabolism could be identified based on co-regulation of the corresponding enzymes. In a recent study, co-regulation analyses of integrated networks identified that cholesterol biosynthesis was highly co-regulated in the liver and interestingly, that mannose metabolism was highly co-regulated in hepatocytes, adipocytes, and myocytes [35]. Mapping the transcriptome of liver and adipose tissue biopsies, co-regulation analyses identified that mannose metabolism in liver tissue was significantly decreased through co-regulation in obese subjects. This finding was corroborated through the analysis of an independent algorithm (reporter metabolite analysis) [36], which identified significant transcriptional changes regarding mannose uptake in the liver. The findings of decreased mannose uptake in liver tissue and subsequent increased plasma mannose levels were validated in 3 independent metabolomics cohorts: (1) lean versus obese subjects (the Relationship between Insulin Sensitivity and Cardiovascular disease [RISC] study); (2) insulin-sensitive versus insulin-resistant obese subjects (the Leipzig study); and (3) high versus low insulin-secreting subjects (the Kuopio study). Interestingly, plasma mannose was significantly enriched in obese, insulin-resistant, and low insulin-secreting subjects, even more significantly than glucose. Furthermore, an independent study of mannose supplementation suggested that mannose could improve insulin tolerance and reduce weight and body fat in mice with high-fat diets if supplemented at early stages [37]. Likewise, integrative network analyses of GEMs could identify potential biomarkers of chronic diseases through multi-omics observations in a comprehensive manner.
A NEW EMERGING PARADIGM: THE HUMAN MICROBIOME
Recently, human microbiome research has been highlighted due to its therapeutic potential beyond the knowledge of lactate-producing bacteria. For example, gut microbiota have been found to (1) successfully cure multiple diseases through transplantation from donors to patients, including Clostridium difficile infection, inflammatory bowel disease, and obesity [38–40]; and (2) modulate the efficacy of host-directed drugs, even immune check-point inhibitors [41,42].
In light of increasing demands for a better understanding of the human microbiome, many approaches have been attempted to reconstruct reference genomes for the human microbiome. However, as many species were unculturable in laboratory conditions, reference genomes for many microbes could not be obtained by in vitro isolations and remained as microbial “dark matter” [43–45]. While powerful, 16S rRNA amplicon sequencing is dependent on reference genomes from isolated bacteria, making it difficult to uncover the whole bacterial population, which includes unculturable species [46]. In addition, this method could introduce biases due to variation in 16S rRNA gene copy numbers, and 16S rRNA sequence conservation limits the ability of this technique to distinguish closely related organisms, posing difficulties for taxonomic classifications at the species and strain levels [46,47].
Recently, computational methods that enable the direct identification of reference genomes from shotgun metagenomics were devised. For example, short reads of shotgun sequencing were assembled and binned into metagenome-assembled genomes (MAGs) based on the similarity of nucleotide compositions [48,49], and co-abundant gene profiling was used to identify clustered gene groups and assemble them into metagenomics species (MGSs), or a metagenomic species pan-genome (MSP) [50,51]. These methods allow us to uncover unculturable species from MAG or MGS/MSP and thereby to quantify the microbiome composition without reference genomes known.
Based on quantitative microbiome profiling of shotgun metagenomics using isolate genomes or MGS, variations of the human microbiome have been associated with diets, diseases, and levels of drug efficacy [52]. For example, a metagenome-wide association study was conducted to investigate the microbial species that are enriched or depleted in adults with diabetes [53], and functional analyses identified that elevated levels of branched amino acids in diabetes were associated with microbial dysbiosis [52]. Unsupervised clustering of human gut microbiome compositions revealed that all individuals can be classified as having three different types of microbiome, called enterotypes, which are associated with different dietary patterns [54]. For instance, the individual gut microbiome can be enriched with Bacteroides, Prevotella, or Firmicutes. Individuals with a high-fat diet and low-grade inflammation were enriched with the Bacteroides type, whereas those with a high-fibre diet were enriched with the Prevotella type. Likewise, the individual microbiome could indicate susceptibility to chronic diseases and predict drug efficacy [41,55], and even could be used in therapeutics to improve metabolic phenotypes, such as obesity [40]. Based on diet-microbiome-host interactions, microbiome studies could guide personalized nutrition for the prevention and early intervention of diseases [56].
CONCLUSIONS
The complex and dynamic nature of human physiology can be studied with systems biology approaches, simplifying complex biological problems into more understandable formulas. Based on quantitative and systems approaches, current healthcare services can be transformed to proactive medicine in the near future. For example, state-of-the-art modelling of human metabolism through GEMs could accelerate the systems understanding of metabolic alterations in diseases by providing computational “maps,” enabling personalized treatments and diets to be suggested. In addition, multi-omics studies with integrative network analyses could also identify which level of omics changes eventually leads to metabolic alterations. For example, metabolic adaptations driving lower levels of mannose consumption in the liver, and subsequently increased plasma mannose levels, were identified. This lack of mannose might lead to insulin resistance because poorly glycosylated hepatic insulin receptors could not clear free insulin in the blood. As a recently emerged axis of human physiology, the human microbiome could explain unexplored variations in human physiology, and quantitative and systems approaches have been devised to study those variations in health and disease.
 XML Download
XML Download