

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
WD40-repeat (WDR) refers to a series of loosely conserved structural motifs comprised of approximately 40 amino acids, often terminating in tryptophan (W)-aspartic acid (D). WDR protein family is a large group of proteins commonly possessing the WDR motifs, that are involved in a wide range of important biological processes. Inherited or acquired defects in WDR proteins result in numerous health problems including neurological diseases, ciliopathies, and cancers. In this review, we provide a unique overview and discussion on the molecular mechanisms and functions of WDR proteins, especially focusing on those that have been associated with human congenital disorders and endocrine diseases. Many of the WDR proteins are called different names mainly due to historical reasons. The official gene nomenclature along with full and alternative names of WDR proteins (based on UniProt) discussed in this review are summarised in the Supplemental Table S1. WDR proteins associated with pathological conditions that are not discussed in the main text in detail are summarised in the Supplemental Table S2.
MOLECULAR STRUCTURE OF WDR PROTEINS
WDR motif was first described in the β-subunit of a GTP-binding protein transducin complex as a sequence of repeats of 40 to 60 amino acids that begin with glycine and histidine (GH) and end with tryptophan and aspartic acid (WD) dipeptides [1]. WDR is an evolutionarily conserved and highly abundant domain in eukaryotes with nearly 1% of human proteomes consisting of WDR-containing proteins [2]. Most recent protein domain database (SMART, http://smart.embl.de/) predicts that 921 WDR proteins are encoded in humans, 591 in Mus musculus and 212 in Drosophila melanogaster. WDR proteins are rarely present in prokaryotes [2]. Each WDR protein can have 4 to 16 copies of WDRs forming seven or more bladed beta-propeller folds [3], which can provide three structural surfaces (top, side, and bottom region of propeller) available to interact with other binding partners [2]. Based on these structural features, it is suggested that WDR proteins could serve as a scaffold that mediates protein-protein or protein-DNA interaction [4]. Since WDRs do not possess any catalytic activity themselves, functional diversity is likely achieved by coordination of multiple binding partners. Mutations in WDR proteins have been reported in several human diseases. Notably, clinically identified mutations of WDR are mostly found on the surface of the protein, presumably interfering their binding interactions with other proteins [1,2,4].
BIOLOGICAL FUNCTIONS OF WDR PROTEINS
WDR proteins can be primarily defined by their sequence similarity in the WD40-repeat domain. However, a wide range of sequence variation has been found in WD40-repeats, resulting in variable numbers of beta-propeller structures. Variations outside of the WD-repeat domain can also contribute to the multidomain contexts [5]. In fact, although all WDR proteins are structurally related, their molecular functions can be quite distinct. This functional diversity is usually acquired from the additional domains present in the respective WDR proteins. Currently, more than 360 additional domains are reported in WDR proteins [6]. The most commonly found additional domains are shown to be functionally involved with ubiquitylation (e.g., F-box [7], SOCS-box [8], RING-finger [9]), microtubule dynamics (e.g., CTLH domain [10]), phospholipid-binding (e.g., FYVE domain [11]) and endocytic vesicle coating (e.g., clathrin terminal domain [12]). WDR domains are also identified in essential subunits of multiprotein complexes that participate in various signalling pathways regulating DNA repair [13-15], protein degradation [16,17], cell cycle control [18,19], mRNA translation [20,21], cilia assembly and maintenance [22,23], and hormone biosynthesis [24]. Therefore, it is not surprising that WDR proteins play important roles in fundamental cellular functions, such as signal transduction, gene expression, RNA processing, protein synthesis, homeostasis, proliferation, apoptosis, intracellular vesicle trafficking, and cargo recognition [2,25-29].
WDR PROTEINS IN HUMAN DISEASE
According to the Online Mendelian Inheritance in Man (OMIM) database (https://omim.org/
), there is a significant correlation between WDR protein dysfunction and human diseases (Fig. 1). Among 360 WDR genes we assessed, 79 genes were reported to be associated with human pathologies which include neurological disorders (40.5%), ciliopathies (21.5%), immune diseases (8.9%), eye problems (7.6%), skeletal anomalies (3.8%), cancers (3.8%), endocrine disorders (2.5%), inflammation (2.5%), and others including preimplantation embryonic lethality (8.9%).
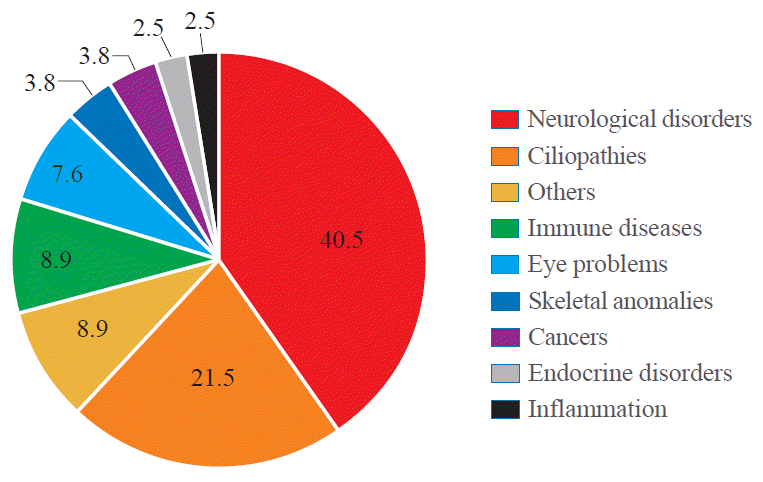
Fig. 1.
A chart indicating the relevant prevalence of human diseases associated with WD40-repeat (WDR) proteins. The data are based on the entries in Online Mendelian Inheritance in Man (OMIM). Total 79 out of 360 WDR proteins have been linked with disease categories as indicated. The ‘others’ category includes multi-organ defects, liver failure, cardiovascular defects and embryonic lethality. The full list of WDR proteins assessed are included in Supplementary Table S1.
![]()
Notably, a significant number of WDR proteins have been associated with ciliopathies, a group of genetic disorders resulting from defects in the structure or function of cilia. Cilia are highly conserved microtubule-based hair-like organelles that extend from the plasma membrane of most vertebrate cells. Cilia can broadly be classified into two types, motile and non-motile (primary) cilia that share the principal axoneme structures [30,31]. The axoneme consists of a circular arrangement of nine pairs of microtubules called outer doublets. In addition to the outer doublets, motile cilia contain a pair of microtubules in the centre called inner doublets [32]. This central pair of microtubules is the scaffold of the central pair complex including radial spokes, inner and outer dynein arms and nexin links [33]. The intraflagellar transport (IFT) particles assemble and maintain the cilium by trafficking ciliary proteins within the cilium [22,23]. Two subcomplexes IFT-A and IFT-B, consisting of at least 6 and 13 proteins, respectively, move along the cilium bidirectionally via retrograde (IFT-A) and anterograde (IFT-B) transport. In retrograde transport (from the ciliary tip to the base), IFT-A uses dynein-2 as a motor, whereas IFT-B is powered by kinesin-2 for anterograde movement [34,35]. Many IFT proteins contain protein-protein interaction motifs including WDRs, tetratricopeptide repeats and coiled coils motifs [22], facilitating the interaction and transport of multiple cargos such as tubulin and dynein components [22,36,37]. Primary cilium serves as a regulatory platform and organising centre for many cellular signalling pathways [38] such as Hedgehog [39], receptor tyrosine kinases [40], and G protein-coupled receptors [41], playing critical roles in normal embryonic development and adult homeostasis [42]. Therefore, defects in the formation and function of primary cilia lead to a wide range of health problems [43], including renal dysfunction, retina degeneration, hypogonadism, diabetes, obesity, hearing impairment, craniofacial/skeletal anomalies, cardiovascular defects, and brain malformations [30,44-46], which are collectively termed as ciliopathies.
WDR proteins associated with neurological disorders
Mutations of WDR proteins are most frequently associated with neurological disorders (see Table 1 for the full list). PAFAH1B1 (LIS1) is the first WDR protein identified in severe brain malformation called lissencephaly type 1 (also known as classic lissencephaly) characterized by the absence or incomplete development of the cerebral cortex, causing unusually smooth brain surface. Lissencephaly can occur in association with other syndromes such as Miller-Dieker syndrome (MDS) [47,48] or as an isolated lissencephaly sequence (ILS) [49,50]. PAFAH1B1 gene is located in 17p 13.3 which is the most frequently deleted chromosomal region in MDS and ILS patients [51]. So far, it is estimated that 65% of ILS patients have deletions or intragenic mutations of PAFAH1B1 [50]. PAFAH1B1 is a microtubule-associated phosphoprotein and its direct interaction with cytoplasmic dynein heavy change is important for neuronal migration, disruptions of which result in lissencephaly [52,53].
Table 1.
List of WDR Proteins Associated with Neurological Disorders
WDR, WD40-repeat; MIM, Mendelian Inheritance in Man; AD, autosomal dominant; AR, autosomal recessive; UV, ultraviolet light; XLR, X-linked recessive; ADHD, attention deficit hyperactivity disorder; CATIFA, cleft lip, cataract, tooth abnormality, impaired intellectual development, facial dysmorphism, attention-deficit hyperactivity; XLD, X-linked dominant.
![]()
Mutations in KATNB1 (LIS6) are associated with complex cerebral malformations known as lissencephaly 6 (microcephaly co-existing with lissencephaly) [54,55]. KATNB1 encodes the p80 subunit of katanin, a microtubule-associated ATPase [56,57] which consists of two subunits [56]. While p60 subunit provides the catalytic function, p80 subunit is the regulatory element that targets this protein to centrosomes and maintains the length of microtubules in developing neurons [58,59].
Mutations in LRRK2 are the most common cause of autosomal dominant Parkinson’s disease [60,61]. To date, more than a hundred mutations of LRRK2 have been identified and six of them are confirmed to be pathogenic [62]. LRRK2 protein binds to the synaptic vesicles and regulates vesicular trafficking by interacting with pre-synaptic proteins such as actin and synapsin [63]. The sequence variation of glycine to arginine at residue 238 (G2385R) which is located between the 5th and 6th WDR domain has been confirmed as a risk factor in the Asian population [64-66]. This mutation modifies LRRK2 protein structure, likely altering its binding affinity to synaptic vesicles and other interactors required for vesicle trafficking [67].
Cockayne syndrome type A (CSA) is a rare neurodegenerative disorder characterized by complex phenotypes including a growth delay, optic atrophy, deafness, abnormalities in limb and digits, and mental disability [15,68]. According to the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ERCC8
), up to 70 mutations of the ERCC8 gene have been reported in CSA so far. ERCC8 is a subunit of E3 ubiquitin ligase complex [13,14] and interacts with ERCC6 during transcription-coupled nucleotide excision repair [13]. ERCC6, a putative helicase, is recruited by stalled RNA polymerase 2 on the DNA damage site and initiates DNA repair by attracting repair proteins including ERCC8 to the lesion [13,15].
Triple-A syndrome (AAAS) is a rare autosomal recessive disorder and patients suffer from adrenal insufficiency, achalasia of the oesophagal cardia, alacrimia, and neurological abnormalities affecting the central, peripheral, and autonomic nervous systems [69,70]. Mutations of ALADIN have been found in all AAAS patients, which results in a truncated protein with loss of function [71]. ALADIN protein is normally localised within nuclear pore complexes [18] but mutants of ALADIN are shown to be sequestered in the cytoplasm [72], leading to impaired nuclear transport of proteins that are required to protect the nucleus from oxidative damage [73,74].
Mutations in WDR4 are reported in patients with microcephaly with severe growth deficiency, seizures, and brain malformations [75,76]. Recent whole-exome sequencing analyses of a family with Galloway-Mowat syndrome (GAMOS) identified a novel mutation of WDR4 [77]. GAMOS present clinically heterogeneous phenotypes which combine renal failure and brain anomalies [78], with additionally associated features including facial dysmorphism, growth retardation and skeletal anomalies [79]. Fibroblast cells derived from patients with GAMOS show defective growth and altered microtubule networks [80]. WDR4 is the human ortholog of yeast Trm82p and forms a complex with N7-methylguanosine tRNA methyltransferase and METTL1, which is essentially required for mRNA translation and stem cell self-renewal and differentiation [20,21]. WDR73 is another WDR protein related to GAMOS [81]. WDR73 is concentrated in the microtubule and interacts with several proteins critical to cell cycle and survival, such as tubulins α/β/γ and Hsp70/ 90 [81].
WDR proteins and cancer predisposition
WDR protein PALB2 is a breast and pancreatic cancer susceptibility factor that interacts with BRCA2 and RAD51C [17,37] facilitating their DNA repair function [82]. Cancer-associated PALB2 mutations cause the loss of its binding ability to BRCA2/RAD51C and biallelic mutations of PALB2 are associated with an increased occurrence of childhood cancers [82].
Another WDR protein FBXW7 is a ubiquitin ligase substrate receptor and the most commonly deregulated ubiquitin/proteasome system (UPS) protein in human cancer [16]. FBXW7 is a tumour suppressor protein that binds to the phosphorylated cyclin E and mediates its degradation by ubiquitination [16,17]. Loss-of-function mutations of FBXW7 result in inappropriate accumulation of cyclin E [17], which is observed in 18% of colorectal cancers, 15% of uterine endometrial carcinoma and 40% of uterine carcinosarcoma [16,17].
WDR proteins associated with ciliopathies
To date, mutations in at least 17 different WDR proteins have been identified in ciliopathies (see below and Table 2). Mutations in all components of IFT-A complex—WDR10/IFT122 [83], WDR19/IFT144 [84], WDR35/IFT121 [85], IFT43 [86], IFT140 [87], TTC21B [88]—and a subset of proteins in IFT-B complex—WDR56/IFT80 [89], IFT172 [90], IFT52 [91], IFT81 [92]—have been identified in skeletal ciliopathies. Cranio-ectodermal dysplasia (CED), also known as Sensenbrenner syndrome, is a ciliopathy characterized by craniofacial and skeletal anomalies [93]. So far, four IFT-A proteins are reported to be mutated in CED, namely, WDR10/IFT122, WDR19/IFT144, WDR35/IFT121, and IFT43 [87,94-97]. Mutations in WDR19/IFT144 have also been identified in patients with Jeune syndrome, also known as asphyxiating thoracic dystrophy (ATD), presenting short stature, short digits (brachydactyly), and respiratory distress due to insufficient rib bone growth [84]. WDR35/ IFT121 mutations are found in both Jeune syndrome and Short-Rib-Polydactyly syndrome (SRPS) [98]. Mutations in WDR34 and WDR60 are also associated with ATD and SRPS [98,99]. WDR34 and WDR60 are subunits of the dynein-2 complex, comprising the two intermediate chains of dynein-2 which mediates retrograde ciliary transport via IFT-A [100,101]. In addition to WDR34 and WDR60, disruptions in other dynein-2 subunits are also common causes of ATD and SRPS [89-91]. Therefore, patients with CED, ATD, and SRPS share clinical and genetic features [102] and can also be affected in non-skeletal organs including kidney, eye, liver, and heart [99].
Table 2.
List of WDR Proteins Associated with Ciliopathies
![]()
AHI1 encodes a protein called Jouberin which contains seven WDR domains [103]. Recessive mutations of AHI1 underlie Joubert syndrome (JS) characterized by abnormal development of brain structures, including the cerebellar vermis and the brainstem, which resemble the cross-section of a molar tooth in MRI, thus nicknamed as ‘molar tooth malformation’ [103,104]. JS patients show additional distinctive features including ocular coloboma, polycystic kidney and polydactyly, which are collectively referred to as JS-related disorders (JSRD) [105,106]. AHI1 mutation is also associated with a broad range of neurological disorders including schizophrenia [107] and autism [108]. Recent mouse model studies have revealed that Ahi1 is highly expressed in the postnatal brain and interacts with other proteins crucial for neuronal differentiation [109,110].
WDR proteins in endocrine disorders
Table 3 lists the WDR proteins involved in endocrine disorders, many of which are often presented as a part of a ciliopathy. Several signalling receptors important in neuro-endocrine functions are shown to localise to primary cilia [111]. They include kisspeptin receptor (KISS1R) [112], type 1 dopaminergic receptor (D1R), beta-2 adrenergic receptor (B2AR) [113], serotonin receptor 6 (5-HT6) [114], and insulin-like growth factor 1 receptor (IGF1R) [115]. It has been suggested that the spatio-temporal distribution and concentration of these receptors on the ciliary membrane surface may provide an additional level of regulation for the signal capacity and specificity of these receptors [116]. Shortening of cilia length and alteration in ciliation frequency can indicate functional disruption of cilia-dependent receptor signalling and protein trafficking, involved in endocrine functions.
Table 3.
List of WDR Proteins Associated with Endocrine Disorders
![]()
WDR11 is a scaffolding protein required for normal ciliogenesis. Mutations of WDR11 have been identified in congenital isolated hypogonadotropic hypogonadism (CHH), septo-optic dysplasia (SOD), combined pituitary hormone deficiency (CPHD), and pituitary stalk interruption syndrome [117-120]. CHH is defined by the absent or delayed puberty due to defective gonadotrophin-releasing hormone secretion or action. CHH can present with a normal sense of smell (normosmic CHH) or defective sense of smell (hyposmic/anosmic CHH or Kallmann syndrome) [121,122]. CHH patients often show other associated features such as midline defects (cleft lip or palate), deformity of hands and feet, neurosensory hearing loss, and ocular motor abnormalities [123-125]. Previous studies have suggested that CHH, SOD, and CPHD are genetically overlapping conditions [120]. Clinically identified mutations of WDR11 caused defective cilia formation, and targeted disruption of WDR11 in animal models resulted in dysgenesis of multiple organs affected in CHH and Kallmann syndrome [126]. Hedgehog signalling pathway which depends on the normal function of primary cilia is also shown to be disrupted by the loss of WDR11. Based on these findings, it was suggested that CHH and Kallmann syndrome could be considered as a ciliopathy [126]. The endocrine feature is also common in other ciliopathies such as JS and JSRD. Some JS/JSRD patients show growth hormone or thyroid hormone deficiency [127], CPHD [128], and micropenis [129].
WDR proteins have been associated with obesity. GNB3 is related to childhood obesity and polymorphism of GNB3 is associated with obesity, hypertension, and diabetes type 2 [130-132]. A genome-wide association study identified WDR11 as a novel genetic locus associated with childhood obesity [133]. Siblings sharing a rare variant of WDR11 gene showed obesity with attention deficit hyperactivity disorder [126]. Obesity is, in fact, one of the main features of Bardet-Biedl syndrome (BBS) [134]. BBS is a ciliopathy with a wide spectrum of clinical features including rod-cone dystrophy, polydactyly, hypogonadism in male and renal abnormalities [135]. Homozygous mutation in WDPCP (also called BBS15) is identified in BBS patients with obesity and male hypogonadism [136]. WDPCP is involved in planar polarity effectors (CPLANE) complex required for recruitment of IFT-A proteins during ciliogenesis [137].
Mutations of TBL1X have been identified in isolated congenital central hypothyroidism [138]. Congenital hypothyroidism (CH) is a thyroid hormone deficiency at birth caused by the impaired function of the thyroid itself (primary CH) or defective stimulation of thyroid gland by a thyroid-stimulating hormone (central or secondary CH) [139]. Central CH can be categorized into two subtypes—isolated thyroid hormone deficiency and CPHD [139]. Isolated thyroid hormone deficiency accounts for 40% of central CH cases [140] and can be caused by mutations in four genes that regulate the thyroid-stimulating hormone biosynthesis, including β subunit of thyroid-stimulating hormone (TSHβ), receptor for thyrotropin-releasing hormone (TRHR), IGSF1 (the regulator for TRHR expression in the pituitary) and TBL1X (an essential subunit of the thyroid hormone receptor corepressor complex) [24,138].
Although AAAS was described as a neurological disorder previously, the adrenal glands are one of the primarily affected organs [71,141]. About 85% of AAAS patients show adrenocorticotropin hormone resistant adrenal insufficiency due to impaired glucocorticoid secretion [142] and a subsequence adrenal androgen deficiency is also observed [143]. Homozygous deletion of DMXL2 is identified in patients with the polyendocrinepolyneuropathy syndrome (PEPNS) [144]. PEPNS refers to a combined symptom including CHH with hypothyroidism, hypoglycemia, peripheral polyneuropathy, and mental disability [143]. A recent study has shown that gonad specific DMXL2 deletion causes impaired spermatogenesis in males [145].
CONCLUSIONS
WDR proteins are widely expressed in human tissues and highly conserved in vertebrates (https://www.proteinatlas.org/search/wdr). Thanks to the recent advancements in genome sequencing analysis, many potentially pathogenic variants of WDR proteins have been identified, which can prove to be powerful tools for assigning new functions to the WDR motifs and associated domains. It is possible that WDR proteins with very similar surfaces have common binding partners or similar functions. Mutations in WDR proteins underlie a broad spectrum of human pathologies including neurological disorders, cancer, ciliopathies, and endocrine disorders. These are complex disorders, thus a clear understanding of the clinical phenotypes and comprehensive diagnosis are often challenging. Molecular mechanisms through which WDR proteins are involved in these diverse conditions remain largely unknown. A better understanding of WDR proteins and their interacting partners may offer some clues. The new insights for WDR-related diseases and their underlying mechanisms as provided in this review may help develop therapeutic approaches targeting the common WDR motifs involved.
 XML Download
XML Download