This article has been
cited by other articles in ScienceCentral.
Abstract
Congenital cystic adenomatoid malformation (CCAM) is a rare developmental dysplastic lesion that affects the fetal bronchial tree. Etiopathogenesis is still poorly understood. Most accepted view is that of abnormal branching of bronchioles during the period of morphogenesis. We observed a rare congenital anomaly of the lungs during fetal autopsy. Routine antenatal ultrasonography revealed multiple echolucent cysts in the right lung of the fetus. Thorough external and internal examination was followed by sectioning of each organ for histopathological examination. Histopathology of the right lung showed distortion of the parenchyma with dilated bronchioles. Multiple cysts lined by columnar epithelium along with loose intervening connective tissue were observed along with many congested and dilated blood vessels. Knowledge of congenital anomalies of the respiratory system would help clinicians to plan the management at a very early stage. Accurate fetal autopsy along with clinical data is important in evaluating fetal deaths and can help in reducing unexplained stillbirths.
Keywords: Congenital, Cystic adenomatoid, Embryology, Lung malformation
Introduction
Congenital cystic adenomatoid malformations (CCAM) are rare cystic lesions of the lung due to developmental anomaly of the lungs. It is characterized by the presence of cystic intrathoracic masses formed by proliferative terminal respiratory bronchioles as well as decrease in the number of normal alveoli [
1]. CCAM affects 1 in 11,000 to 1 in 35,000 live births [
2], with male predominance [
3]. CCAM was first reported by Chin and Tang in 1949 [
4], and later, Stocker et al. [
5] in 1977 gave its classification. We report a rare case of unilateral cystic malformation of the right lung of a 24-week fetus during autopsy. We anticipate that with this case report, we will be able to gain a better understanding of this congenital defect and can increase awareness about its importance.
Case Report
A 30-year-old multigravida, at 24 weeks of gestation with gravida 3, parity 1, abortion 1 and live birth 1 (G3P1A1L1) with a previous history of intra uterine death due to some congenital anomaly (not known to the parents), presented to the obstetrics outpatient department for a routine antenatal visit. Routine ultrasound examination revealed a large cystic lesion within the right chest along with ventriculomegaly and ascites. The left lung was found to be normal. The fetal diaphragm and stomach were found in normal positions. The most likely diagnosis made was CCAM of the right lung. Due to the high possibility of pulmonary hypoplasia and nonimmune hydrops fetalis, the parents intended a medical termination of the pregnancy.
Following termination of pregnancy, a male fetus of weight 750-gram was received, along with an attached placenta and umbilical cord for autopsy, that was conducted at the Department of Pathology, ESIC Super Specialty Hospital in collaboration with the Department of Anatomy, ESIC Medical College and Hospital at Hyderabad. The fetus was then kept in 10% formalin, with a small niche given on the scalp and abdomen for effective perfusion into all spaces. External inspection revealed no anomalies. An “I” shaped incision was given, followed by complete evisceration. All the organs were carefully observed for any gross anomalies and then processed for histopathological examination (
Fig. 1A). While all the organs were grossly normal, the right lung was observed to be flattened with distorted architecture and presence of multiple small cysts measuring less than 1 cm on the posterior aspect (
Fig. 1B, C).
Histopathology revealed distorted lung parenchyma with dilated bronchioles. Multiple small cystic spaces were lined by simple columnar epithelium along with loose intervening connective tissue and the presence of congested and dilated blood vessels (
Fig. 2). Other viscera were found normal according to gestational age. With the above given findings, a diagnosis of CCAM Stocker Type III was suggested.
Discussion
CCAM is predominantly unilateral, affecting only one lobe of the lung. Left-sided CCAMs account for the bulk of antenatally diagnosed CCAMs [
6]. Data is minimal on bilateral CCAMs as there are very less number of case series reported [
6]. Bilateral lesions may be associated with an inherited predisposition to an underlying cell signaling abnormality, which may increase the likelihood of eventual malignant transformation in the CCAM or elsewhere in the lung [
7] Prenatally diagnosed CCAMs have been linked to a wide range of perinatal mortality rates, ranging from 9% to 49% [
6]. We tried to compare the findings which are listed in
Table 1 [
8-
11].
Etiopathogenesis: embryological and genetic basis
CCAMs are hamartomatous lesions that can affect any part of the fetal respiratory system, starting from the trachea to the alveoli. As the etiopathogenesis of CCAM is still unclear, various theories have been suggested, which include abnormal tissue proliferation [
4], hyperplasia, dysplasia or metaplasia. There are four distinct phases of vertebrate lung development and maturation based on the anatomical changes that occur in lung architecture: pseudo-glandular (5–16 weeks), canalicular (16–26 weeks), saccular (26–36 weeks), and alveolar (36 weeks to maturity) (
Table 2) [
12]. Studies suggest that the majority of CCAMs occurs during the pseudo glandular stage. The bronchial type of epithelium is arrested at this phase of lung development, resulting in CCAM disease types I–III, whereas the alveolar acinar type of epithelium is arrested later in weeks 22–36, resulting in CCAM pathology type IV [
9].
The presence of normal alveoli on the periphery of the lesion but abnormal growth in the middle, implies failure of terminal bronchiole to canalize during second trimester, resulting a discontinuity of the conducting and the respiratory elements [
13]. Bronchoscopic and microscopic studies revealed that bronchial atresia is the primary defect that contributed to the development of CCAM [
14]. Between 5 and 16 weeks of pregnancy, the pseudoglandular, or branching phase of lung development occurs. It involves a plethora of spatially and temporally controlled signaling molecules that allow broad communication between the pulmonary epithelium and the mesenchyme.
HOXB5, which affects lung growth during branching morphogenesis, is significant gene that may influence the development of CCAM. Its expression is kept at a level that is appropriate for early lung development. In comparison to normal fetal lung or term CCAM tissue, CCAM that expanded faster, progressed to hydrops fetalis, and required in utero resection exhibited increased platelet derived growth factor (
PDGF-B) gene expression and PDGF-BB protein synthesis [
15].
FABP-7, a novel pulmonary gene has been described, expression of which was found to be lower in fetal CCAM than in control fetal lung at both the RNA and protein levels, suggesting that it may play a role in lung development and CCAM pathogenesis [
15]. Thus, elaborate research on the
FABP-7 gene can give more insight into the pathogenesis of CCAM.
Classification of CCAM
Stocker in 1977, was the first to classify CCAM, based on the number and size of the cyst and histopathological findings [
2,
5]. CCAM classification schemes have changed over time, and there are now five primary categories based on the embryologic stage of genesis and histologic features that are presented in
Table 3 and
Fig. 3. According to this classification, we report a case of Type III CCAM.
Anomalies associated with CCAM
CCAM has been linked to other congenital anomalies such as cardiovascular malformations, lung hypoplasia, congenital diaphragmatic hernia, extralobular sequestration, hydrocephalus, bilateral renal agenesia/dysgenesis, gastrointestinal atresia, polyhydramnios, mediastinal shift, and particularly nonimmune fetal hydrops [
5]. In the present case, no anomaly was observed, but antenatal ultrasonography revealed ascites, which could indicate the onset of hydrops.
Conclusion
Because lung disease is a prevalent cause of fetal death, clinicians should be aware of congenital defects of the respiratory system so that treatment can be planned at an early stage. CCAM is most commonly diagnosed in the antenatal or neonatal period. Serial ultrasonographic evaluation, fetal lung mass size, and fetal echocardiography are required for antenatal diagnosis of cases. To achieve the best results, very attentive monitoring during the perinatal period is required. Accurate fetal autopsy combined with clinical data is critical, and it can help reduce unexplained stillbirths.
Acknowledgements
We would like to express our gratitude to the parents of the fetus who chose to donate their fetus to the pathology department for research.
References
1. Kanavi JV, Thangaraj J, Thomas A. 2017; Congenital cystic adenomatoid malformation (CCAM): antenatal and postnatal management. Int J Reprod Contracept Obstet Gynecol. 6:2447–51. DOI:
10.18203/2320-1770.ijrcog20172329.

2. Stocker JT. Stocker JT, Dehner LP, editors. 2001. The respiratory tract. Pediatric Pathology. Lippincott Williams & Wilkins;Philadelphia: p. 445–517. DOI:
10.1201/9781315382531-36.
4. Chin KY, Tang MY. 1949; Congenital adenomatoid malformation of one lobe of a lung with general anasarca. Arch Pathol (Chic). 48:221–9. PMID:
18137795.
5. Stocker JT, Madewell JE, Drake RM. 1977; Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 8:155–71. DOI:
10.1016/S0046-8177(77)80078-6. PMID:
856714.
6. Davenport M, Warne SA, Cacciaguerra S, Patel S, Greenough A, Nicolaides K. 2004; Current outcome of antenally diagnosed cystic lung disease. J Pediatr Surg. 39:549–56. DOI:
10.1016/j.jpedsurg.2003.12.021. PMID:
15065026.
7. Cacciari A, Ceccarelli PL, Pilu GL, Bianchini MA, Mordenti M, Gabrielli S, Milano V, Zanetti G, Pigna A, Gentili A. 1997; A series of 17 cases of congenital cystic adenomatoid malformation of the lung: management and outcome. Eur J Pediatr Surg. 7:84–9. DOI:
10.1055/s-2008-1071060. PMID:
9165253.
8. Chikkannaiah P, Kangle R, Hawal M. 2013; Congenital cystic adenomatoid malformation of lung: report of two cases with review of literature. Lung India. 30:215–8. DOI:
10.4103/0970-2113.116272. PMID:
24049259. PMCID:
PMC3775204.
9. Patil AG, Karangadan S, Kishore V. 2015; Congenital cystic adenomatoid malformation of lung in fetus: report of two cases with brief review of literature. J Assoc Chest Physicians. 3:53–6. DOI:
10.4103/2320-8775.158854.
10. Annam V, Korishetty SI, Yelikar BR, Hippargi SB, Shivalingappa DB. 2010; Bilateral congenital cystic adenomatoid malformation, stocker type III with associated findings and review of literature. Indian J Pathol Microbiol. 53:331–3. DOI:
10.4103/0377-4929.64324. PMID:
20551549.
11. Boulot P, Pages A, Deschamps F, Hedon B, Laffargue F, Viala JL. 1991; Early prenatal diagnosis of congenital cystic adenomatoid malformation of the lung (Stocker's type I); a case report. Eur J Obstet Gynecol Reprod Biol. 41:159–62. DOI:
10.1016/0028-2243(91)90094-2.
12. Sadler TW. Sadler TW, editor. 2012. Respiratory system. Langman's Medical Embryology. 12th ed. Lippincott Williams & Wilkins;Philadelphia: p. 202–6. DOI:
10.5005/jp/books/10252_13.
13. Ostör AG, Fortune DW. 1978; Congenital cystic adenomatoid malformation of the lung. Am J Clin Pathol. 70:595–604. DOI:
10.1093/ajcp/70.4.595. PMID:
360825.
14. Moerman P, Fryns JP, Vandenberghe K, Devlieger H, Lauweryns JM. 1992; Pathogenesis of congenital cystic adenomatoid malformation of the lung. Histopathology. 21:315–21. DOI:
10.1111/j.1365-2559.1992.tb00401.x. PMID:
1398534.
15. Liechty KW, Crombleholme TM, Quinn TM, Cass DL, Flake AW, Adzick NS. 1999; Elevated platelet-derived growth factor-B in congenital cystic adenomatoid malformations requiring fetal resection. J Pediatr Surg. 34:805–9. discussion 809–10. DOI:
10.1016/S0022-3468(99)90377-9. PMID:
10359185.
Fig. 1
(A) Male fetus (gestational age, 24 weeks) received for autopsy. ‘I’ shaped nick given followed by opening of thoracic and abdominal cavity. (B) En bloc of lung tissue removed during autopsy, showing multiple cysts (red arrow). (C) Formalin fixed lung tissue of fetus, showing cystic wall (red arrow).
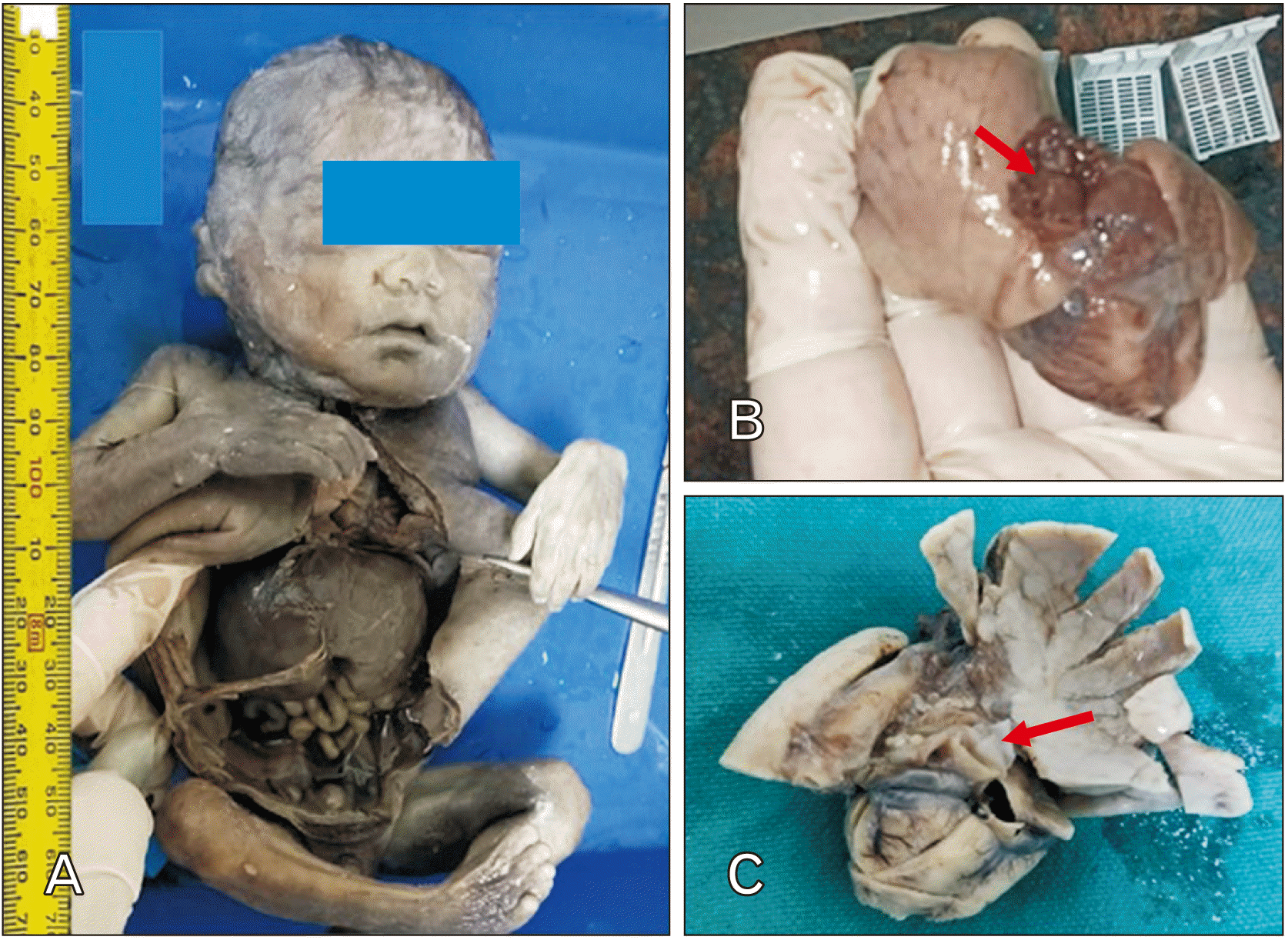
Fig. 2
(A) Section from lung tissue showing multiple small cystic spaces (CS) with adjacent alveoli (H&E, ×100), (B): Higher magnification image of the cyst showing cyst wall lined by columnar epithelium (red arrows) (H&E, ×400). BV, blood vessel.
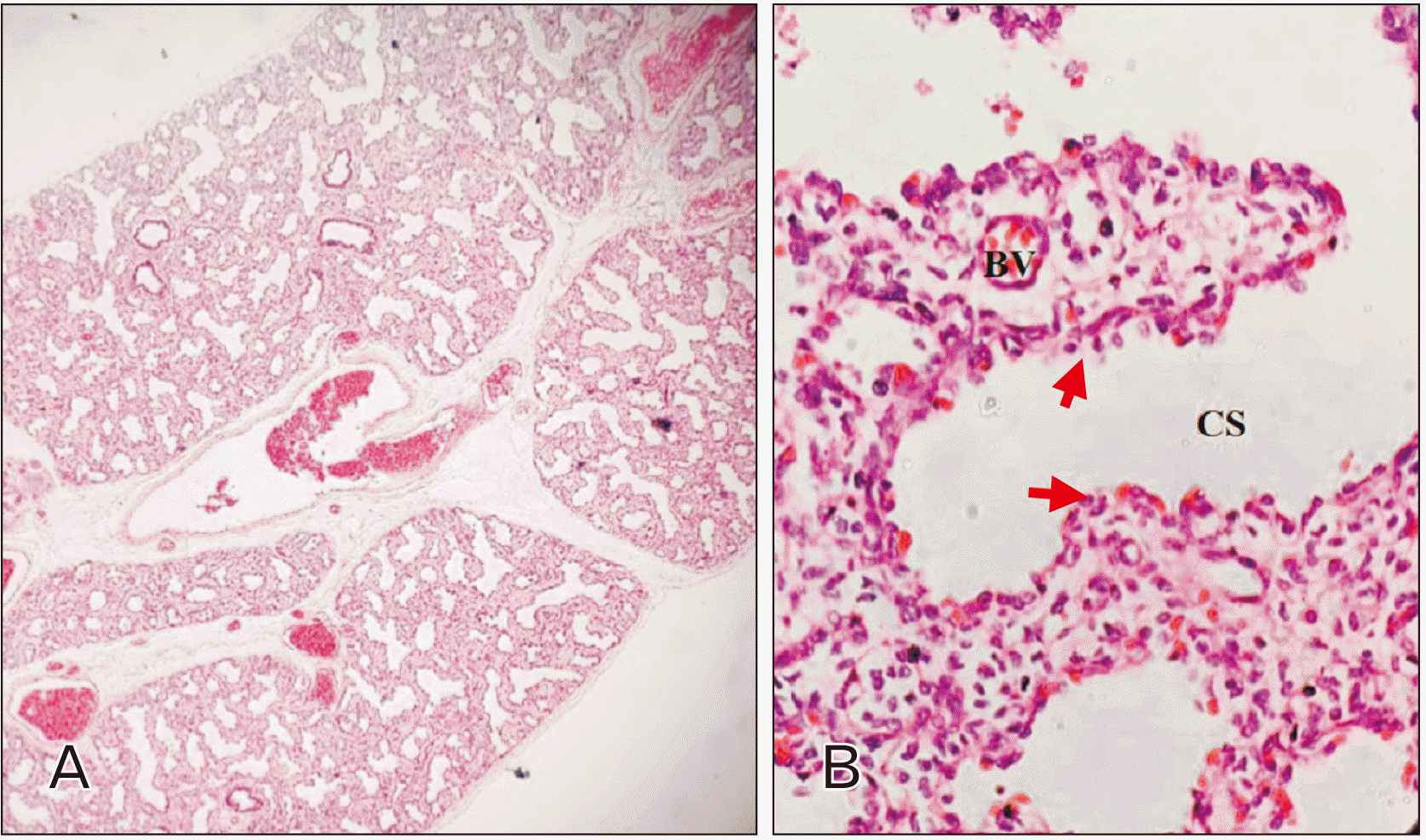
Fig. 3
Showing classification of congenital cystic adenomatoid malformation (CCAM) according to site of origin in the tracheobronchial tree (type 0–4). Data from Stocker et al. (Hum Pathol 1977;8:155-71) [
5].
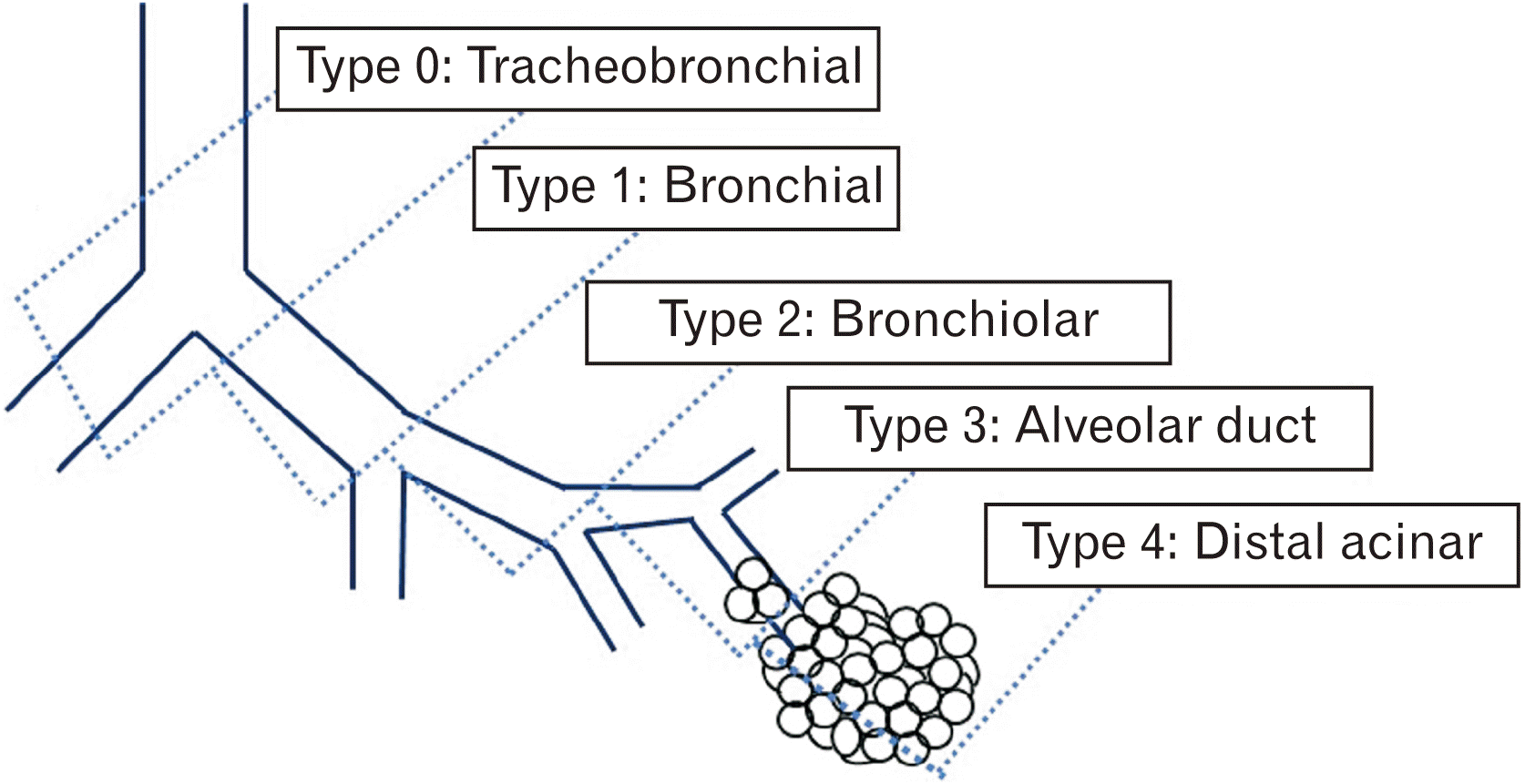
Table 1
Comparison of classical autopsy findings observed in congenital cystic adenomatoid malformation (CCAM) in other studies
|
Author |
GA at the time of diagnosis (wk) |
Sex and weight of the fetus |
Lung involved |
Characteristics of the cyst |
Lining epithelium |
Classification (Stocker) |
|
Chikkannaiah et al. [8] |
20.5 (Case 1) |
Male, 1.45 kg |
Right lung was enlarged, left lung was normal |
Multiple cysts of size <2 cm |
Stratified columnar epithelium, and presence of thin fibromuscular septa |
CCAM Stocker type II |
|
24.5 (Case 2) |
Female, weight not mentioned |
Left lung was involved, right lung was hypoplastic |
Multiple cysts of size <2 cm |
Lined by flattened cuboidal epithelial cells having fibromuscular septa |
CCAM Stocker type I |
|
Patil et al. [9] |
20 (Case 1) |
Male, 500 g |
Right lung was involved, displacing the cardia towards left |
2.7×2.5 cm cystic in posterior aspect of right lung |
Pseudostratified columnar epithelium with thin fibromuscular septa |
CCAM Stocker type I |
|
40 (Case 2) |
Female, 2.5 kg |
Left lung was involved |
Two cystic lesions 2×2 cm and 2×1 cm respectively |
Flattened alveolar type epithelial cells |
CCAM Stocker type IV |
|
Annam et al. [10] |
16 |
Male, weight not mentioned |
Bilateral involvement of lungs, displaced heart |
Lungs appeared solid without obvious cysts
Cut section showed multiple minute cysts less than 2 mm in diameter |
Ciliated cuboidal epithelium, and sequestered by numerous alveoli sized cystic structures lined by non-ciliated cuboidal epithelium |
CCAM Stocker type III |
|
Boulot et al. [11] |
20 |
Male, 400 g |
Enlarged right lung with left mediastinal shift |
Multiple cysts were seen |
Ciliated cuboidal or cylindrical epithelium |
CCAM Stocker type I |
|
Present study |
24 |
Male, 750 g |
Right lung was distorted and flattened |
Multiple small cysts each of <1-cm size |
Simple columnar epithelium along with loose intervening connective tissue, congested and dilated blood vessels |
CCAM Stocker type III |
Table 2
Different phases of normal development and maturation of vertebrate lung [
12]
|
Development period |
Maturation of lung |
|
Weeks of gestation |
Developmental changes |
|
Pseudo-glandular period |
5–16 weeks |
|
|
Canalicular period |
16–26 weeks |
Respiratory bronchioles are formed from terminal bronchioles Respiratory bronchiole divides into Alveolar ducts Lung is well vascularised
|
|
Terminal sac period |
26–36 weeks |
|
|
Alveolar period |
36 weeks to childhood |
|
Table 3
Classification of congenital cystic adenomatoid malformation (CCAM) of lung according to Stocker et al. [
5]
|
Type of CCAM |
Incidence |
Origin |
Size of the cyst |
Epithelium |
Associated abnormality |
|
Type 0 |
Rarest type, but severe and usually lethal |
Trachea or bronchus |
Small |
|
Mostly not associated with anomalies |
|
Type 1 |
Most common (50% to 70% of cases) |
Distal bronchus or proximal bronchiole |
Large cysts but lesser in number
Sometimes a single dominant cyst may also be present |
Usually lined by ciliated pseudostratified epithelium, wall of cyst is thin usually |
Because these CCAMs may be large, they may have significant mass effect, which can lead to hydrops |
|
Type 2 |
15% to 30% of cases |
Terminal bronchioles |
Smaller cysts, usually 0.5 to 2 cm, solid areas may be present |
Ciliated cuboidal or columnar epithelium |
Most associated with other anomalies |
|
Type 3 |
5% to 10% of cases |
Acinar part |
Multiple small cysts, appearance as solid mass, sometimes large enough to distort the thoracic contents |
Ciliated cuboidal or columnar |
|
|
Type 4 |
5% to 15% of cases |
Alveolar |
Large cysts as large as 10 cm |
Alveolar lining cells |
Mostly associated with malignancy, specifically pleuropulmonary blastoma |


 PDF
PDF Citation
Citation Print
Print



 XML Download
XML Download