

 PDF
PDF Citation
Citation Print
Print



Introduction
Cell migration is a complex process in embryonic development and homeostasis. In gastrulation, the migration of all cells is necessary for the formation of three embryonic layers, namely endoderm, mesoderm, and ectoderm. Also, in the expansion stage of neurons during embryonic development, migration from the conical head of neuronal cells is evident. However, the migration process is not limited to the developmental step; rather, it can occur in adults both in physiological and pathological conditions [1, 2]. Most cells in multicellular organisms can move during specific phases of tissue formation, maintenance, repair, and immune responses. Cell movement is also essential in the development of some diseases, such as neuroinflammation or various types of cancer [3]. The force of cell movement is primarily defined by the interaction between cells and the extracellular matrix (ECM) or other cells. The molecular organization of interactions and functions are comparative and vary among cells and tissue types [1, 4, 5]. Most of our knowledge about cell migration has been obtained from in vitro studies on two-dimensional (2D) substrates, which has provided new insights into numerous basic cell migration mechanisms, interacting with substrates and changing the speed and direction of movements on 2D surfaces. The biology of cells surrounded by the ECM in the living native environment of organisms differs from when they are cultured on a 2D surface [6]. When cells are exposed to the 2D surface, they develop dorsoventral polarity, which does not occur in in vivo conditions [7]. Besides, the ability of cells to move in a three-dimensional (3D) ECM depends on the viscosity and hardness of the ECM [8]. Therefore, the analysis of cell movement and migration in a 3D environment could further mimick the cell environment in an in vivo condition [5]. The established in vitro cell culture models allow direct analysis of physicochemical parameters of cell migration, including the role of the ECM dimension, stiffness, and functional tissue barriers, as well as the subsequent single or collective cell migration [1, 4]. In vivo models, such as embryonic cells of drosophila, zebrafish, or mouse, help us to understand the role of the ECM in cell migration under normal physiological or even pathological states [3, 6, 9]. The ECM creates scaffolds as a support for cells in all tissues and organs, comprising of glycoproteins, fibronectin (FN), laminin, proteoglycans, and non-matrix proteins, such as growth factors [10, 11]. The ECM-derived scaffolds are made by decellularization of natural tissues, eliminating the impact of cellular antigens and, at the same time, preserving the structural and functional proteins of the ECM. The ECM-derived scaffolds provide a 3D environment for cell behaviors, including growth, polarity, and differentiation [10]. Also, the evaluation of cell migration and dynamic tissue culture could pave the way to understand molecular and cellular mechanisms underlying tissue repair in multicellular organisms. Blastema tissue is one of the types of tissues created during the repair process [12, 13]. It contains a group of undifferentiated cells possessing the ability to proliferate, differentiate, and migrate [11-13]. Previous studies have shown that organisms that develop blastema tissue could be attractive experimental living models to investigate the process of differentiation, development, and repair. New Zealand rabbit pinna, which can form blastema tissue after making a hole in it, can be a useful experimental model for tissue regeneration and repair process [11, 12].
FN is one of the macromolecules involved in cell migration. FN is found in both soluble and insoluble forms that the latter form is present in the ECM. The matrix of FN provides migration pathways during development and facilitates the migration and proliferation of fibroblasts during tissue healing [7, 14, 15]. Tenascin C (TNC) is a highly conserved glycoprotein amongst vertebrates usually found in the ECM [16, 17]. TNC binds to different extracellular binding molecules, such as FN, collagen, fibrillin-2, glypican, perlecan, and periostin [18]. TNC can also attach to different viral and bacterial pathogens [18, 19]. TNC is synthesized in various sites of developing embryonic tissues; however, it has restricted distribution in adult stem cell niches, wound healing, or in pathological conditions, including cancer and inflammation [18, 20-22]. In the present study, after creating a decellularized bovine mesenteric tissue using a combination of physical and chemical protocols, blastema tissue was created from the rabbit pinna in the presence of FN. The interaction and migration process of blastema cells with the ECM of the bovine mesentery was also evaluated histologically, and the expression of the TNC gene was assessed.
Go to : 
Materials and Methods
Preparation of ECM‐derived mesenteric scaffold
The entire experimental procedures of the current research were confirmed by the Ethics Committee of Ferdowsi University of Mashhad (ethical code: IR.UM.REC.1398.122). In this experimental study, bovine mesenteric tissue was harvested from the animals immediately after being sacrificed. The mesenteric tissues were cut into cubic‐like pieces with a dimension of 5 mm×5 mm. Mesenteric tissues were stored at –4°C before decellularization. Frozen samples were then thawed at room temperature and washed with sterile normal saline. Then, the specimens were treated with a 1% sodium dodecyl sulfate (SDS) solution (Merck, Germany) for 24 hours and next placed in 1% Triton X-100 solution for 12 hours, at 37°C, accompanied with gentle agitation.
After decellularization, the remaining SDS and Triton X-100 were removed from the ECM‐derived mesenteric scaffold and sterilized. Afterward, the specimens were rinsed in the sterile phosphate-buffered saline (PBS) buffer for 4 hours in a shaker incubator. Then, the samples were washed with 70% ethanol for 2 hours. In hygienic conditions, the scaffolds were washed with sterile distilled water and then rinsed again in sterile PBS solution for 1 hour. Finally, to prepare the scaffolds for cell culture, they were incubated in Dulbecco’s modified eagle’s medium (DMEM; Gibco, Brooklyn, NY, USA) for 4 hours in a 5% CO2 incubator.
Preparation of blastema tissue
In order to prepare blastema tissue, four New Zealand white male rabbits were purchased from Razi Vaccine and Serum Research Institute, Mashhad, Iran, with an age range of 3–6 months and weight of 2–2.5 kg. The animals were kept under laboratory conditions. First off, the hairs existing around the rabbit pinna were removed with hair removal cream and then locally anesthetized using a lidocaine solution. Then, three holes with a diameter of 4 mm were created in the ear using a punching apparatus. Two days after punching the ears, the blastema ring appeared in the periphery of the primary hole in the pinna of the ears. The punched area was isolated and placed in normal saline.
Assembly and culture of ECM‐derived mesenteric scaffold with blastema tissue
The blastema tissue ring was washed twice in PBS; then, the ECM‐derived from mesenteric tissue was placed into the blastema tissue ring. These assemblies were immediately placed in a 24-well plate containing DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 µl penicillin/streptomycin (Sigma‐Aldrich, Taufkirchen, Germany), and 100 µg/ml Fibronectin (human; Invitrogen, Carlsbad, CA, USA). The assemblies were incubated at 37°C in a 5% CO2 atmosphere. All samples were subjected to histological staining and RT-PCR on days 7, 10, 15, and 21 after the cell culture process.
Histological analysis
All samples were fixed in Bouin’s solution. After fixation, specimens were dehydrated by ascending gradient of ethanol and then embedded in paraffin. The praffinized specimens were sectioned at a thickness of 7 μm using a microtome (Leits, Vienna, Austria). Next, the paraffin-embedded samples were deparaffinized by xylene, rehydrated, and stained appropriately. In order to determine cellularity, hematoxylin and eosin (H&E) staining was used. Masson’s trichrome (Merck, Darmstadt, Germany) staining was performed to identify the cellular and ECM structures. Also, 4, 6 diamidino‐2‐phenylindole (DAPI; Sigma‐Aldrich) staining was also used to assess nuclear localization.
Cell number assay and statistical analysis
In order to determine the cell numbers migrated to target tissue, we assayed this factor at four different time points, including days 3, 10, 15, and 21. For this purpose, on each desired day, the cell numbers from different zones of at least four stained slides with H&E are counted and the mean of cell numbers in each slide is calculated. The statistical analysis was performed using GraphPad Prism (version 8) software. The Kruskal–Wallis test and Dunn’s multiple comparison test are used to determine the statistically significant difference between different time points. Significant differences between groups were considered at P≤0.05.
RNA isolation and RT-PCR
The RNA content of samples, including the ECM‐derived mesenteric scaffold assembled with blastema tissue, were extracted using a Total RNA purification Kit (Jena Bioscience, Jena, Germany). Afterward, the extracted RNA was transcribed into complementary DNA (cDNA) using random hexamer primers using AccuPower RT PreMix (Bioneer, Daejeon, Korea). Briefly, 250 ng of the total RNA samples were added to each AccuPower CycleScript RT PreMix (dN6) tube, then filled up to 20 µl with DEPC water. The lyophilized pellet was dissolved by taping and spin down methods. The obtained cDNA was amplified at 20°C for 30 seconds, 43°C for 4 minutes, and 55°C for 30 seconds, and this cycle was repeated 12 times. The heat-inactivation of the RT enzyme was performed at 95°C for 5 minutes. The PCR amplification was carried out using Phusion High-Fidelity PCR Kit (Thermo Scientific, Waltham, MA, USA) with the following thermocycling conditions; 98°C for 30 seconds, followed by 33 cycles (98°C for 10 seconds, 60°C for 20 seconds, and 72°C for 20 seconds/kb) and a final extension step of 7 minutes at 72°C. The PCR reaction was conducted at a final volume of 20 µl, including 2 µl cDNA, 1 µl forward and reverse primers (10 µM), 0.4 µl dNTPs (10 µM), 4 µl Phusion HF buffer (5X), 0.6 µl DMSO, 0.2 µl Phusion DNA Polymerase, and 14.8 µl distilledwater (dH2O). PCR primers used in this study were designed according to the consensus sequences of the TNC genes obtained from the GenBank database of the National Center for Biotechnology Information and using AlleleID software (version 7). The primers are listed in Table 1. Eventually, PCR products were subjected to electrophoresis with 1% agarose gel.
Go to :
Results
Characterization of the decellularized mesenteric tissue by histological analysis
Mesenteric tissue was decellularized using the physicochemical technique. The effects of this method on the obtained decellularized mesenteric tissue were evaluated histologically. H&E staining of native and decellularized tissue revealed complete decellularization of mesenteric tissue. Masson’s trichrome staining also indicated that collagen distribution in the ECM. The results demonstrated a qualitative decellularization process and appeared a decellularized mesenteric ECM accompanied by the elimination of cells and preservation of collagen fibers (Fig. 1).
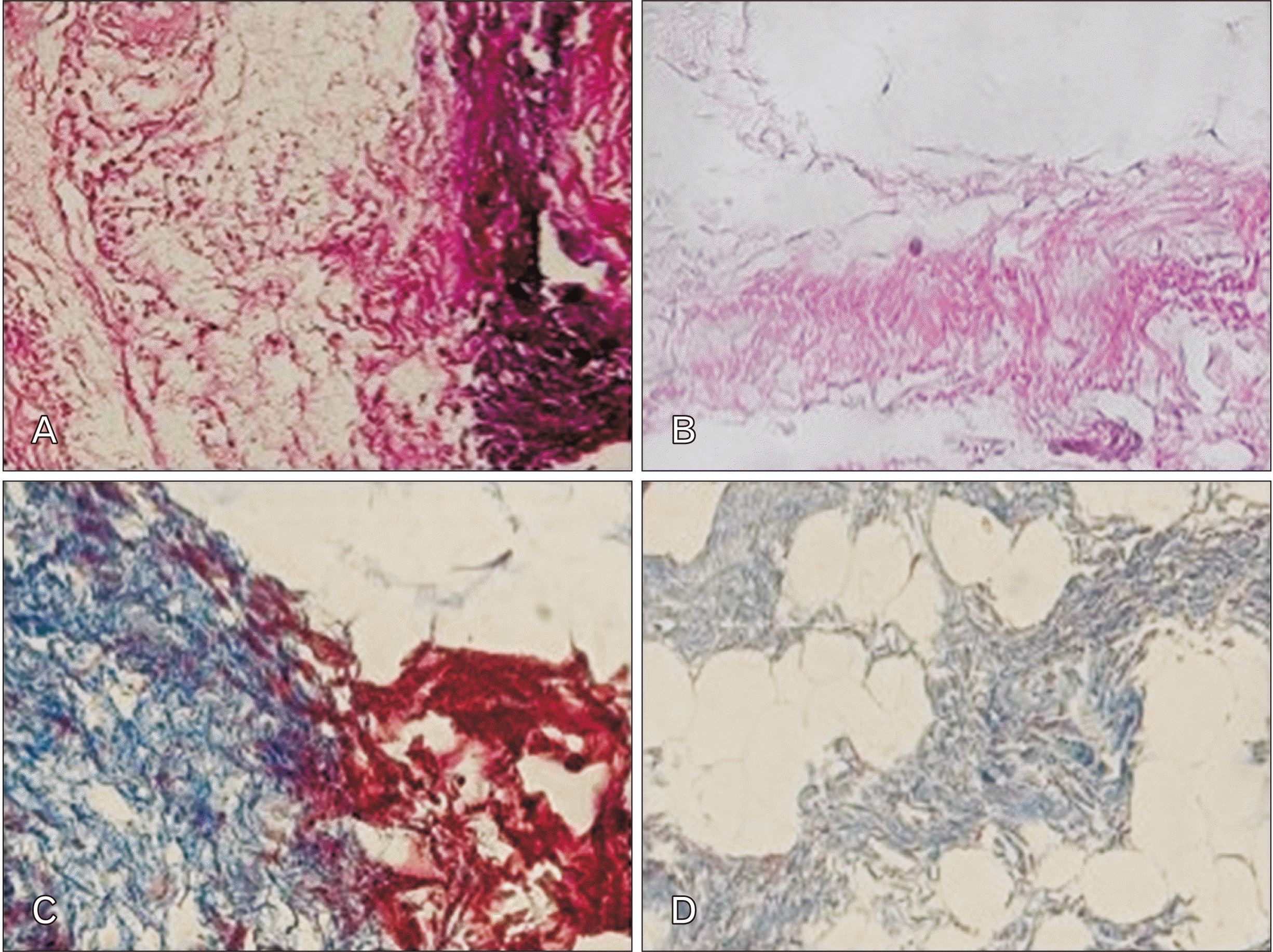 | Fig. 1Control and decellularized mesenteric samples after H&E and Masson's trichrome staining methods. (A, B) The construct of the decellularized mesenteric is displayed in comparison with the native mesenteric tissue using H&E staining. (C, D) Masson’s trichrome staining demonstrated that in decellularized mesenteric ECM, the components remained intact. Magnifications for A, B, C, and D are ×200. ECM, extracellular matrix.
|
Investigation of interactions between Blastema tissue and decellularized mesenteric tissue
After sterilization, the decellularized mesenteric matrix was assembled in blastema tissue rings derived from rabbit pinna to investigate the interaction in the presence of FN factor. A schematic overview of all the stages to achieve the assembled speciments of the ECM-scaffold is shown in Fig. 2. The assembled specimens were subjected to histological staining on different days.
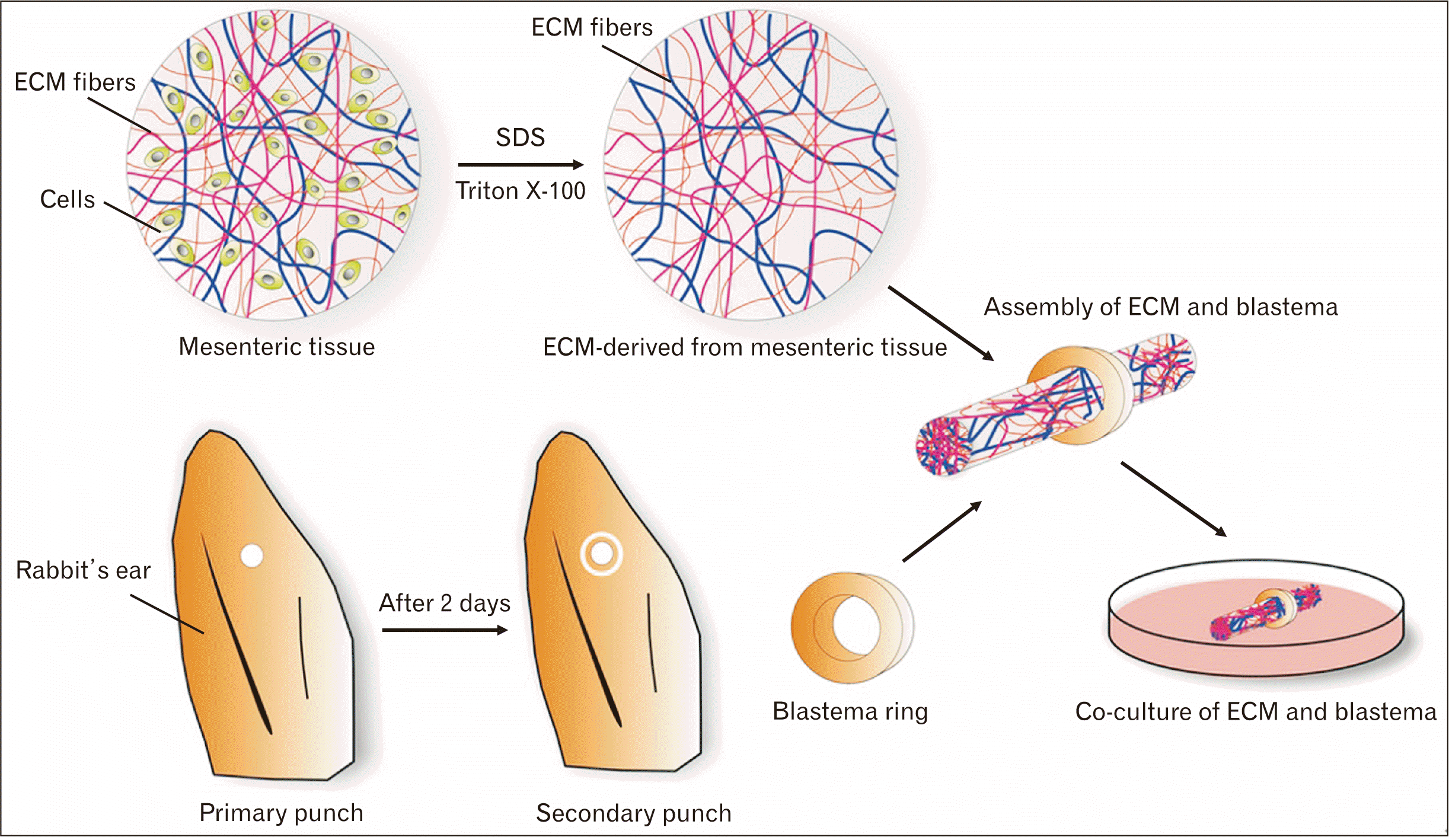 | Fig. 2Schematic overview of the preparation, assembly, and co-culture of the mesenteric ECM and blastema tissue ring. SDS and triton X-100 solutions decellularize the mesenteric tissue. As well as, to prepare blastema tissue, the New Zealand rabbit’s ears were punched, and two days after punching the ears, the blastema ring derived from the periphery of the primary hole. Next, the blastema rings and the ECM scaffolds were assembled and co-cultured in a medium containing fibronectin. ECM, extracellular matrix; SDS, sodium dodecyl sulfate.
|
H&E staining of sections obtained from the assembled specimens showed that only mesenteric decellularized matrix was evident, and the cells were not detectable at the region of the interaction three days after the cell culture (Fig. 3A, B); however, ten days after the cell culture process, blastema cells were distributed in the margin of the mesenteric decellularized matrix. On days 15 and 21, a merked number of cells were visible in the outer regions of blastema tissue, and the surrounding of the decellularized mesenteric matrix was well detectable. Also, the polarity of the cells towards the matrix, as well as relatively elongated cell morphology, was detected (Fig. 3).
 | Fig. 3H&E staining. (A, B) Three days after the cell culture, only mesenteric decellularized tissue was observed (magnification: A, ×100 and B, ×200). (C, D) Cells on day 10 at the site of interaction between blastema tissue and mesenteric extracellular matrix (ECM) are observed (magnification: C, ×200 and D, ×400). (E, F) The cell polarity is shown at the site of interaction between blastema tissue and the mesenteric matrix on day 15 (magnification: E, ×200 and F, ×400). (G, H) Cell migration indicates blastemal cells, along with the mesenteric matrix and proliferating cells at this site on day 21 (magnification: G, ×200 and H, ×400). Arrows show adherence and maintenance of blastema cells in the ECM scaffold during culture after the 10th day.
|
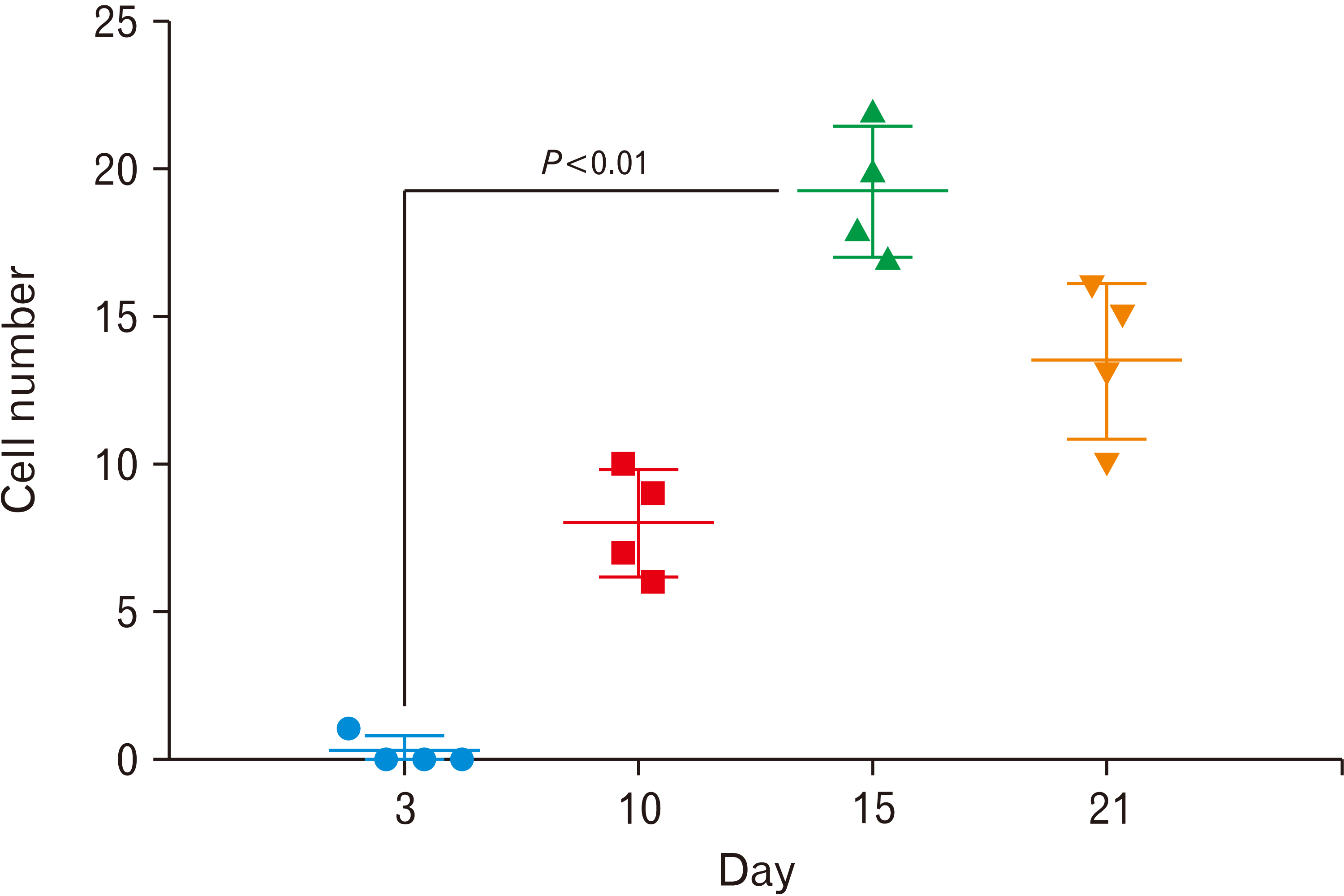
The adhesion and polarity of blastema cells in the decellularized mesenteric matrix were also analyzed after Masson’s trichrome staining. In this staining, collagen is stained in blue, nuclei are stained in dark purple, and cytoplasm is stained in light purple colores. Three days following the cell culture process, according to hematoxylin-eosin staining, only decellularized mesenteric matrix was observed (Fig. 4A, B). After ten days of the cell culture, the cells were well migrated onto the decellularized mesenteric matrix, while, on days 15 and 21, cell penetration was evident in peripheral areas and within the collagen fibers of the matrix (Fig. 4). Specific quasi-vessel structures with spindle-specific polarity, as well as cell division at the margin of the mesenteric decellularized matrix, are shown in Fig. 4G. Moreover, the number of cells that migrate into the ECM scaffolds was assayed on days 3, 10, 15, and 21. The results of counting the penetrating cells at different time points clearly revealed that the highest cell number was achieved on day-15 after culture, which was significant compared to day-3 (P≤0.01) (Fig. 5).
 | Fig. 4Masson’s trichrome staining. (A, B) On day 3, only mesenteric decellularized tissue was evident (magnification: A, ×100 and B, ×200). (C, D) Cell proliferation in blastema tissue, especially in the peripheral areas on day 10 of the cell culture (magnification: C, ×100 and D, ×200). (E, F) On day 15, the polar structures of cells are detectable at the point of the interaction between blastema tissue and the mesenteric matrix (magnification: E, ×200 and F, ×400). (G, H) Blastema cells are observed along with the mesenteric matrix with vascular-like structures on day 21 (magnification: G, ×200 and H, ×400). Arrows blastema cells’ attachment and penetration in the marginal zone of the extracellular matrix scaffold since the 10th day after culture.
|
The native and decellularized mesenteric tissues and the assembled specimens on day 15 of the cell culture were examined by DAPI staining and evaluated under a fluorescence microscope. Native bovine mesenteric tissue that was depicted with bright nuclei indicates the presence of intact cells in mesenteric tissue. There were no signs of cells in the decellularized tissue that proved the efficiency of the decellularization procedure. On day 15 of the cell culture process, blastema cells were detected as bright spots migrating toward the ECM derived from mesenteric tissue (Fig. 6).
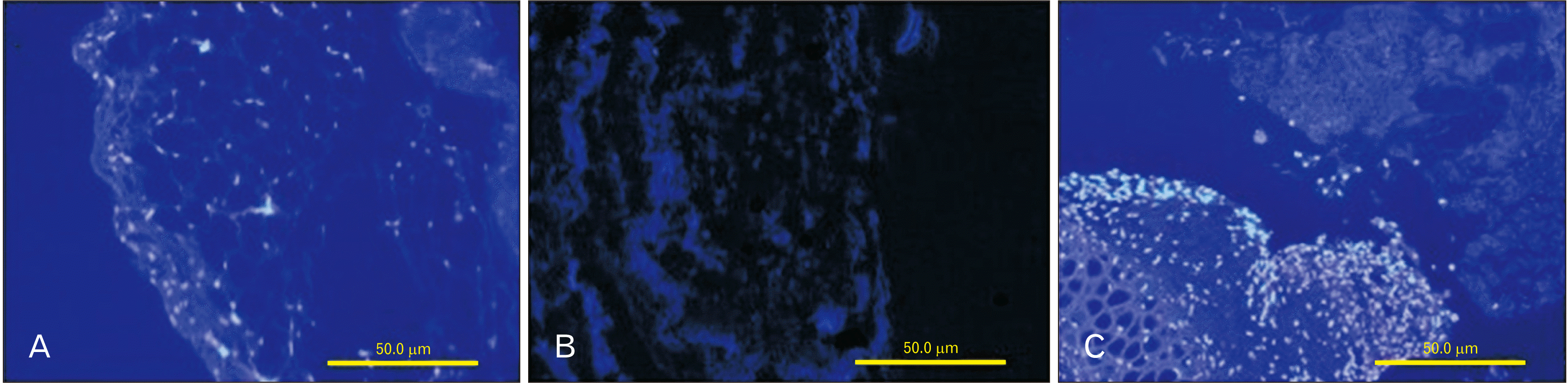 | Fig. 6Staining with DAPI before and after decellularization. Native bovine mesenteric tissue shown in bright nuclei represents the presence of intact cells in native tissue (A). The elimination of cell nuclei in obtained decellularized mesenteric compared with native mesenteric tissue (B). On day 15, blastema cells were depicted as bright spots in the interaction site with the decellularized mesenteric tissue (C). Magnifications for A, B, and C are ×200. DAPI, 4,6 diamidino-2-phenylindole.
|
TNC can be involved in some biological processes, including cell migration. In order to analyze whether TNC is expressed in assembled specimens, the TNC expression was assessed using RT-PCR. The results showed that TNC expression was markedly enhanced when blastema cells had the most penetration and migration into decellularized mesenteric tissue on days 15 and 21 of the cell culture (Fig. 7).
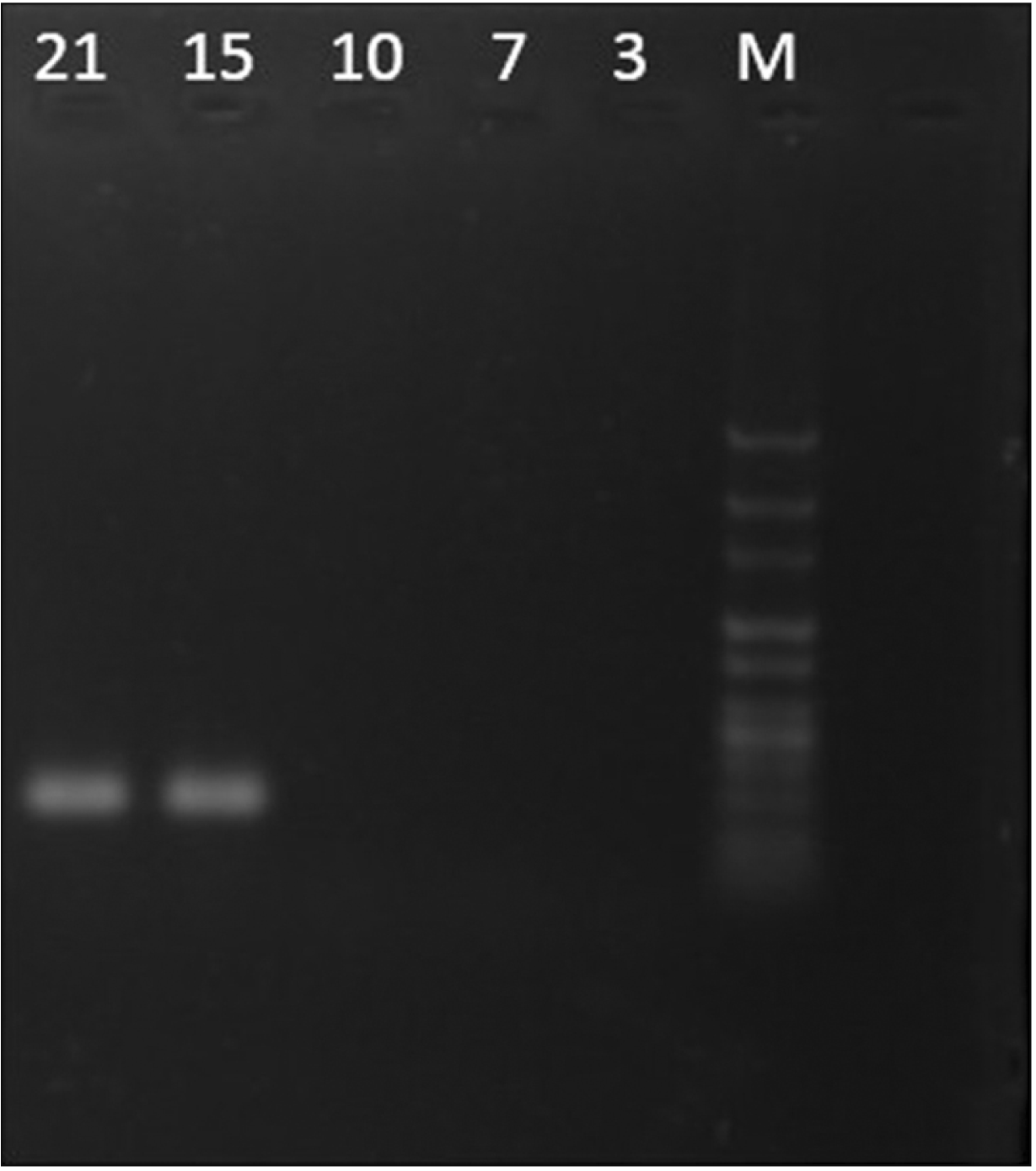
Go to :
Discussion
The use of empirical reality-based models in cell migration studies can be significant and valuable. Generally, our ability to understand cellular interactions with the ECM is derived from two-dimensional cell culture or the analysis of animal models. On the other hand, animal models, which are often used in final tests, do not have enough ability to reproduce different aspects of the cell migration process, tumor metastasis, drug response, autoimmune diseases, and stem cell differentiation [23, 24]. Therefore, the 3D cell culture in vitro can provide a similar cell culture environment to animal models [25, 26]. After punching the rabbit pinna, the growth of the epithelial tissue is converged on repairing the hole in vivo [13, 27]. In this study, a decellularized mesenteric matrix in the presence of FN was used to evaluate the interaction with blastema tissue. First, to prepare the ECM with the characteristics of an environment with fibrous molecules, lack of antigenic effect, and cell adhesion, the natural decellularized bovine mesenteric tissue was established using the physicochemical method. Various decellularization methods have been developed for several animal tissues such as blood vessels [28], heart valves [29], submucosal layer of small intestine [30], skin [31], tendons and ligaments [32], urinary bladder [10], bone [33], and cartilage [34]. Previous studies showed that snap freeze‐thaw cycles followed by treatment with effective detergents, such as SDS and Triton X-100, can lead to the removal of cellular components from the tissues, without any effect on collagen content and other ECM components. H&E staining demonstrated the removal of cells after the decellularization process in bovine mesenteric tissue (Fig. 1). Also, DAPI staining confirmed the complete elimination of DNA from the decellularized mesenteric tissue (Fig. 6A, B).
The decellularized mesenteric tissue is a suitable candidate for the interaction of cells with the ECM, as well as polarity and cell migration, during cell culture due to the maintenance of microstructures of the matrix and suitable interconnected structures. The decellularized mesenteric placed into the blastema tissue, which was derived from rabbit pinna, was used to assess the interaction among these structures at different days in an environment containing FN. FN has been used as a coating for scaffold materials to support host biocompatibility. So, it is an attractive and more cell-friendly substrate than collagen or laminin protein for in vitro cell culture. FN has a tripeptide Arg-Gly-Asp (RGD) motif that is essential for cell adhesion through the integrin. In previous studies, it has also been shown that FN-rich pathways guide and increase the migration of many types of cells during wound healing and embryonic development [35, 36]. The study on the third day after the cell culture showed that no cell penetration into the decellularized mesenteric matrix. However, on day 10 of the cell culture, the presence of cells at the boundary between blastema tissue and decellularized mesenteric matrix was evident. At longer culture periods, on days 15 and 21, the penetration of blastema cells into the decellularized mesenteric matrix was detectable (Figs. 3, 4). These observations were confirmed by counting the penetrating cells into the decellularized mesenteric matrix, which showed the highest number of migrated cells belonged to days 15 and 21 after culture (Fig. 5).
Blastema tissue is typically found during the development of the embryo and tissue regeneration [12, 13]. This tissue contains a mass of undifferentiated cells that could grow, migrate, and differentiate into various tissues and organs [11, 13]. TNC has been reported to be associated with migrating cells into embryo tissues, wound healing, or metastasis of cancer cells [17, 18]. We evaluated the TNC expression in the assembled specimens on different days and found that the expression of this gene was observed only on days 15 and 21 of the cell culture when the cells entirely migrated into the matrix (Fig. 7). In the present research, the functional interplay between the TNC gene and FN was analyzed in an experimental model of interaction between blastema cells and the ECM derived from bovine mesenteric tissue. Since TNC is a distinguished migration-associated matrix component with the anti-adhesive activity, it may control blastema cell migration into the matrix [37]. Also, we used a medium containing FN that might be involved in the polarity and adhesion of blastema cells to the matrix.
These findings show that the ECM-derived mesenteric tissue prepared by the physicochemical decellularization method can support cells polarity and adhesion. This model can serve as a 3D microenvironment for biological processes due to the tendency and migration of blastema cells to the ECM-derived mesenteric tissue.
Go to :
 XML Download
XML Download