

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Second-generation antipsychotics (atypical antipsychotics) are the most widely used treatments for psychosis, particularly in schizophrenia. First-generation antipsychotics (typical antipsychotics) primarily inhibit dopamine type 2 (D2) receptors in the nervous system, whereas the second-generation antipsychotics inhibit several neurotransmitters, such as serotonin (5-hydroxytryptamine1A, 2A, 2C, 3, 6, and 7) and norepinephrine (α1 and α2) receptors, including D2 receptors [1,2]. Numerous second-generation antipsychotics have been developed, including clozapine, olanzapine, asenapine, quetiapine, iloperidone, risperidone, ziprasidone, lurasidone, and aripiprazole. Clozapine is a relatively early second-generation antipsychotic, but it has excellent clinical efficacy in the treatment of schizophrenia. In fact, while ~30% of cases of schizophrenia are resistant to other second-generation antipsychotics, clozapine effectively improves clinical symptoms in patients with its superior therapeutic efficacy [3]. Despite its clinical efficacy, several side effects have been reported from clozapine, the most common of which is agranulocytosis. Treatment with clozapine can also cause sedation, hypersalivation, tachycardia, hypotension, hypertension, and weight gain [4,5]. Clozapine can also have side effects on ion channels. For example, clozapine was reported to inhibit recombinant Cav3.1 T-type calcium channels in human embryonic kidney-293 cells [6]. Clozapine was also shown to inhibit human ether-a-go-go related gene (hERG) K+ channels, causing prolongation of the QT interval [7]. Cardiovascular adverse effects are commonly observed in patients treated with atypical antipsychotic drugs. Although the mechanisms underlying these cardiovascular abnormalities have not been elucidated, inhibition of hERG channels can lead to these cardiovascular abnormalities. Indeed, use of clozapine is associated with the development of myocarditis and cardiomyopathy [8,9]. To date, however, the effects of clozapine on vascular ion channels have not been studied. Therefore, we investigated the effects of clozapine on vascular K+ channels, specifically voltage-dependent K+ (Kv) channels, using freshly isolated coronary arterial smooth muscle cells.
Vascular K+ channels mainly regulate resting membrane potential, and therefore vascular tone. Several types of K+ channel have been identified in vascular smooth muscle cells. Among these, Kv channels are important for the regulation of resting membrane potential and are mainly activated by membrane depolarization [10-12]. Indeed, inhibition of Kv channels by 4-aminopyridine induces vasoconstriction in rabbit aortic smooth muscle [13]. Furthermore, several reports have suggested that alterations or defects of vascular Kv channel function are closely related to various cardiovascular pathological conditions including hypertension, diabetes, and atherosclerosis [14,15]. Therefore, the side effects of some drugs on vascular Kv channels should be elucidated to avoid misunderstanding the results of vasculature studies.
In the present study, we demonstrated the inhibitory effects of clozapine on Kv channels using native rabbit coronary arterial smooth muscle cells. Our findings suggest that clozapine inhibits vascular Kv channels in a concentration- and use (state)-dependent manner independent of its serotonin and dopamine receptor antagonistic effects.
Go to : 
METHODS
Animals and single smooth muscle cell preparation
Male New Zealand White rabbits (7–9 weeks old, weighing 1.8–2.3 kg) were purchased from Nara Biotech (Seoul, Korea). All experiments were performed in accordance with the guidelines of the Committee for Animal Experiments, Kangwon National University (approval No. KW-210512-1). The rabbits were anesthetized by intravenous injection of sodium pentobarbital (45 mg/kg) with heparin (100 U/kg) to minimize pain. The hearts were removed and placed in normal Tyrode’s (NT) solution, and the coronary artery was dissected out from the left side of the ventricular septum and cleaned of connective tissue, fat, and blood. The separation process was performed by immersion in NT solution at room temperature (20°C–22°C) under a stereomicroscope. Before enzymatic treatment, the lumen of the artery was cut open to enhance the effectiveness of the enzymatic process. The artery was first rinsed with digestion medium (Ca2+-free NT solution) containing 1.0 mg/ml dithiothreitol (DTT), 1.1 mg/ml bovine serum albumin (BSA), and 1.15 mg/ml papain for 19–22 min at 37°C. Subsequently, the artery was transferred to next digestion medium containing 1.0 mg/ml DTT, 1.1 mg/ml BSA, and 3.0 mg/ml collagenase for 16–18 min at 37°C. Single smooth muscle cell suspensions were obtained by trituration 15–30 times in Kraft-Brühe (KB) solution with fire-polished 9-inch Pasteur pipettes, and these acute dissociated cells were maintained at 4°C in KB solution and used within 1 day.
Solutions and drugs
The NT solution, which is extracellular solution in patch-clamp experiment, had the following composition (in mM): 133 NaCl, 5.2 KCl, 16 glucose, 0.5 NaH2PO4, 6 HEPES, 0.6 MgCl2, and 1.7 CaCl2 (pH 7.4 adjusted with NaOH). The pipette (internal) solution contained (in mM): 111 K-aspartate, 22 KCl, 9.5 HEPES, 4.4 NaCl, 1.3 MgCl2, 5 Mg-ATP, and 10 EGTA (pH 7.25 adjusted with KOH). The KB solution contained (in mM): 51 KCl, 68 KOH, 23 KH2PO4, 3.2 MgCl2 9.5 HEPES, 0.5 EGTA, 17 Taurine, 47 L-glutamate, and 16 Glucose (pH 7.35 adjusted with KOH). DPO-1, guangxitoxin, and linopirdine were purchased from Tocris Cookson (Ellisville, MO, USA) and clozapine was purchased from Sigma Chemical Co. (St. Louis, MO, USA). All drugs were prepared as stock solutions in distilled water or dimethyl sulfoxide (DMSO), and were diluted in daily bath (external) solution. The concentration of DMSO in the final dilution did not exceed 0.1% and did not affect the Kv current at this concentration.
Electrophysiological recordings
Acute dissociated cells were placed in a constant flow bath containing NT solution on the stage of an inverted microscope at room temperature (20°C–22°C). The Kv currents were recorded using an EPC-8 amplifier (Medical System Corp., Darmstadt, Germany) and NI-DAQ-7 interface (National Instruments, Union, CA, USA) under voltage-clamp conditions. Patchpro software was used to acquire data and generate voltage pulses. Data were digitized at 2 kHz with filtering at 1 kHz. Patch electrodes were fabricated from borosilicate glass capillaries (1.5 mm outer diameter × 1.17 mm internal diameter × 75 mm length) using a Narishige PP-830 puller (Narishige Scientific Instrument, Tokyo, Japan). The tip resistance of the patch electrodes containing the pipette solution was 2–3 MΩ. The series resistance (7.18 ± 0.35 MΩ, n = 22) was continuously monitored during Kv current recording. If the series resistance was increased, data were not used. Input resistance was 2.33 ± 0.18 GΩ (n = 20) under control conditions, and was not significantly altered by application of clozapine (2.35 ± 0.19 GΩ, n = 20). The average cell capacitance was 14.22 ± 0.71 pF (n = 18). All electrophysiological experiments were performed at room temperature.
Data fitting and statistics
The results were analyzed using OriginPro 8 software (Microcal Software, Inc., Northampton, MA, USA). The half-inhibitory concentration (IC50) and Hill coefficient (n) were determined from concentration–response results using the Hill equation:
where ƒ is the percentage inhibition of the Kv current (ƒ = [1 − Idrug/Icontrol] × 100%) at each test potential and [D] is the clozapine concentration.
Steady-state activation curves were determined from the tail currents obtained by returning the potential to −40 mV after short (20–40 ms) depolarizing pulses from −80 to +60 mV. The activation curves were fitted with the Boltzmann equation:
where V, V1/2, and k represent the test potential, the half-point of activation, and the gradient value, respectively.
Steady-state inactivation was obtained using a two-step voltage protocol. The currents were elicited by returning the potential to +40 mV after preconditioning prepulses (7 s) of −80 to +30 mV. The inactivation curves were fitted to another Boltzmann equation:
where V, V1/2, and k indicate the test potential, half-point of inactivation, and the gradient value, respectively.
All data are given as the mean ± standard error of the mean (SEM). Statistical significance was assessed at the 95% confidence level (p < 0.05) using the Mann–Whitney U-test. Significant differences are indicated in graphs by asterisks.
Go to :
RESULTS
Clozapine-induced inhibition of Kv currents
The inhibitory effect of clozapine on steady-state Kv currents was investigated using the conventional patch clamp technique. To exclude other vascular K+ channels such as large conductance calcium-activated potassium (BKCa) and ATP-sensitive potassium (KATP) channels, 10 mM EGTA and 5 mM ATP were included in the pipette solution, and 100 nM iberiotoxin and 10 µM glibenclamide were applied to the bath solution. Furthermore, the Inward-rectifier potassium (Kir) channels were also excluded by using conduit coronary arteries, which do not include these channels [16]. Kv currents were evoked using step depolarizing pulses to +60 mV from a holding potential of −80 mV in steps of 10 mV. Fig. 1A presents typical Kv currents from coronary arterial smooth muscle cells. The recorded Kv currents peaked rapidly and then slowly inactivated during depolarization. Application of 10 µM clozapine to the external solution decreased the Kv currents within 2 min. Fig. 1C presents the current–voltage (I–V) relationships of control Kv currents and clozapine-induced inhibition of Kv currents measured at the end of the pulse. Clozapine-induced inhibition of steady-state currents was observed in the full voltage range over which Kv channels were activated. For example, 10 µM clozapine inhibited Kv currents by 40% at 60 mV.
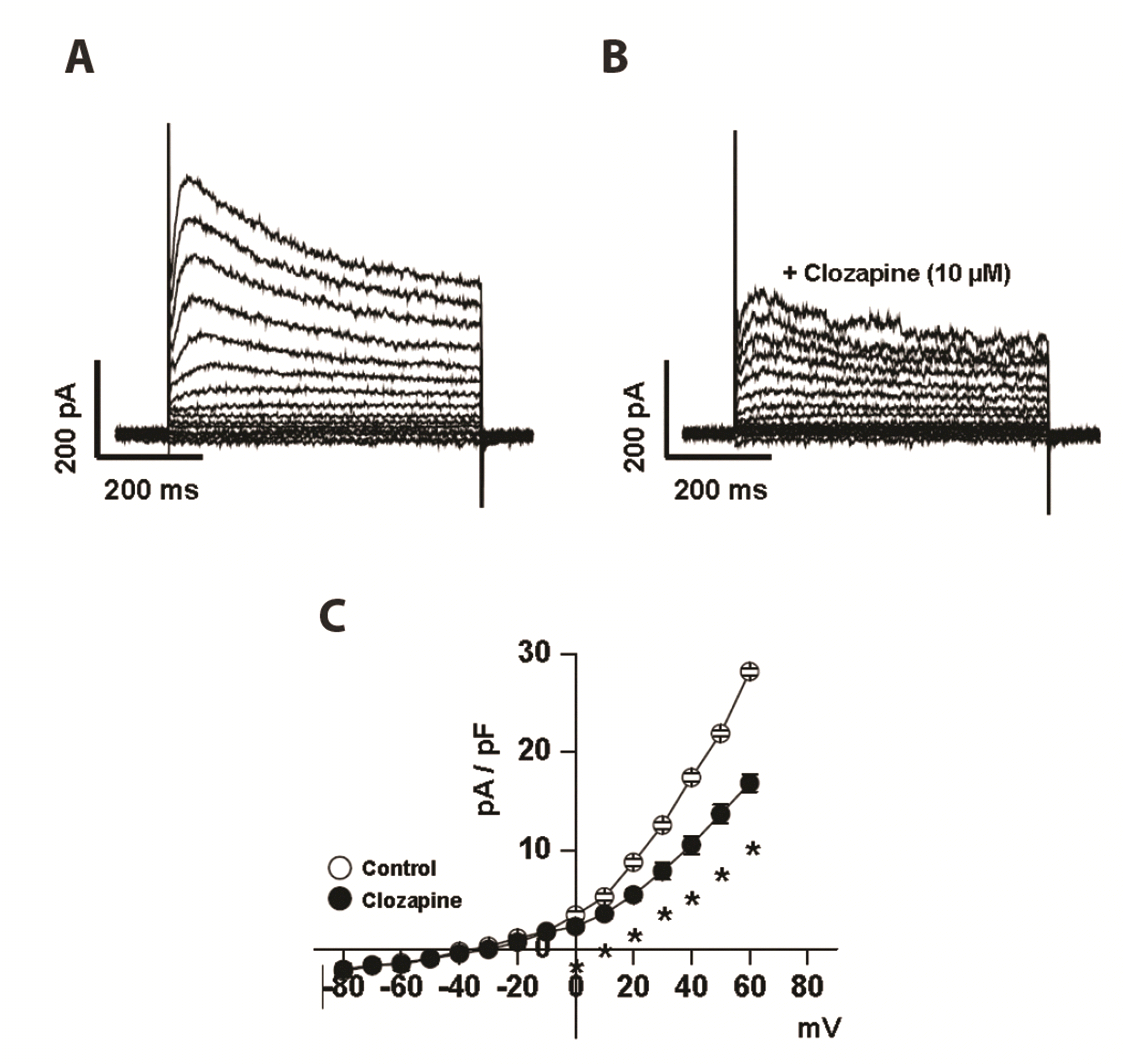 | Fig. 1Inhibitory effect of clozapine on voltage-dependent K+ (Kv) currents in rabbit coronary artery smooth muscle cells.Kv current traces were obtained in response to 600-ms step-depolarizing pulses from a holding potential of −80 mV in the (A) absence or (B) presence of 10 μM clozapine using the same cells. (C) The current–voltage (I–V) curves of the steady-state Kv currents in the absence (○) and presence (●) of 10 μM clozapine. n = 8. The “n” means the number of cells. Only one cell was used from a rabbit to reduce individual differences. Data are mean ± SEM. *p < 0.05 (control vs. clozapine, at each voltage).
|
Concentration dependence of clozapine-induced inhibition of Kv currents
The concentration dependence of clozapine-induced inhibition of steady-state Kv channels was investigated using various concentrations of clozapine (0.01, 0.03, 0.1, 1, 3, 10, 30, 50, 100, and 300 µM). Each concentration was applied in a cumulative manner for at least 2 min to reach a steady-state of Kv current inhibition. The Kv currents were elicited by applying one-step depolarizing pulses to +60 mV from the holding potential of −80 mV. As shown in Fig. 2A, increasing concentrations of clozapine augmented inhibition of the Kv current. Data were fitted to a Hill equation, yielding an IC50 value of 7.84 ± 4.86 µM and a Hill coefficient of 0.47 ± 0.06. These results indicate that clozapine inhibited the Kv currents in a concentration-dependent manner. The higher concentration of 100 µM clozapine did not induce further inhibition of Kv currents, suggesting that the maximal concentration of clozapine to inhibit Kv currents is 100 µM.
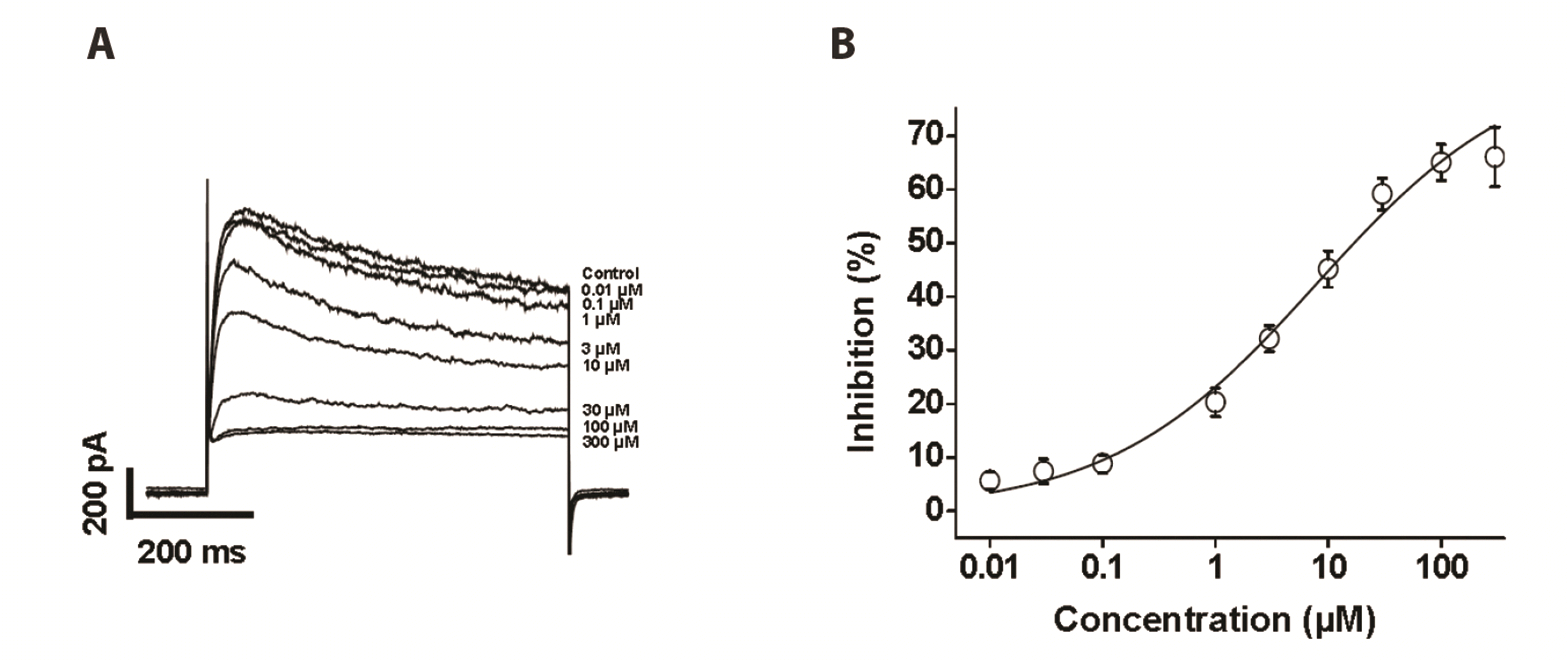 | Fig. 2Concentration dependence of the effects of clozapine on voltage-dependent K+ (Kv) currents.Current recordings of Kv channels were elicited by 600-ms one-step depolarizing pulses of +60 mV from a holding potential of −80 mV. (A) Superimposed current traces of Kv currents with increasing concentration of clozapine. (B) Concentration dependence of the inhibitory effect of clozapine. The inhibition (%) of Kv currents measured at the end of pulses is plotted against the clozapine concentration. The smooth line shows the best fit with a Hill equation. n = 11. Data are mean ± SEM.
|
Effects of clozapine on the steady-state activation and inactivation kinetics of Kv currents
The voltage dependence of steady-state activation and inactivation was investigated to examine whether the clozapine-induced inhibition of Kv currents was due to changes in activation and inactivation kinetics. The activation curves of Kv currents in the absence and presence of clozapine were obtained from the tail currents and were fit to a Boltzmann equation as described in the Materials and Methods section. As shown as Fig. 3A, the activation curve of Kv currents was unaffected by 10 µM clozapine. The half-point of activation (V1/2) and gradient value (k) were −8.18 ± 1.37 mV and 12.02 ± 0.99 under control conditions, and −6.84 ± 2.65 mV and 11.01 ± 1.65 in the presence of 10 µM clozapine, respectively.
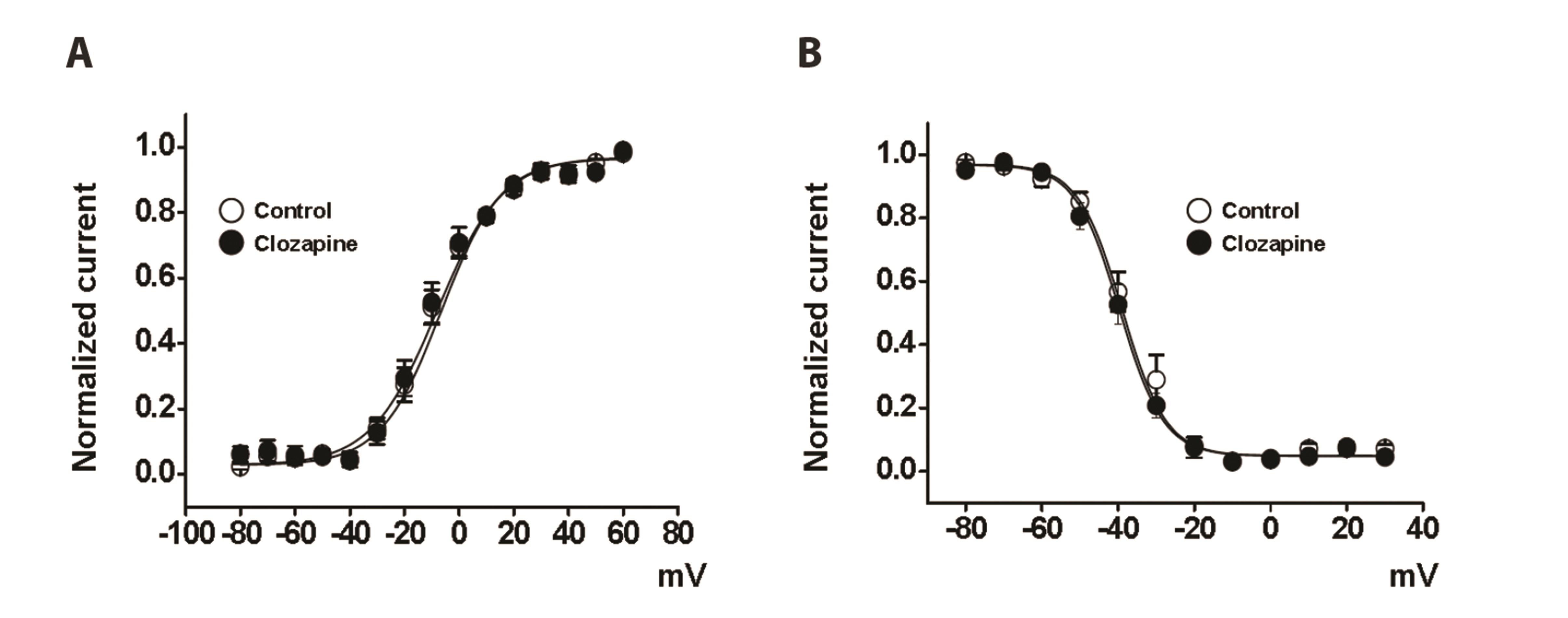 | Fig. 3Lack of effect of clozapine on voltage dependence of steady-state activation and inactivation curve.(A) Activation curves of the voltage-dependent K+ (Kv) currents in the absence (○) and presence (●) of 10 μM clozapine. The tail currents were evoked by a returning potential of −40 mV after 20–50 ms short depolarizing pulses at different voltages. n = 9. (B) Inactivation curves of the Kv currents in the absence (○) and presence (●) of 10 μM clozapine. The curves were elicited by a returning potential of +40 mV after 7-sec depolarizing pulses at different voltages. n = 9. Data are mean ± SEM.
|
The steady-state inactivation curves were obtained from the currents elicited by applying a two-step voltage protocol and fit to another Boltzmann equation as described in the Materials and Methods section. The application of 10 µM clozapine did not affect the inactivation curve (Fig. 3B). The half-point of inactivation (V1/2) and gradient value (k) were −38.88 ± 1.56 mV and 5.77 ± 1.02 under control conditions, and −39.61 ± 1.11 mV and 5.78 ± 0.70 in the presence of 10 µM clozapine, respectively. These results suggest that clozapine did not affect the gating properties of Kv channels.
Use (state) dependence of clozapine response on the Kv currents
The use (state) dependence of clozapine-induced Kv channel inhibition was examined by applying 20 repetitive depolarizing pulses of +60 mV from a holding potential of −80 mV at frequencies of 1 and 2 Hz. The peak amplitudes of Kv currents at each pulse were normalized by the peak amplitude of the first Kv current, and then plotted against the pulse numbers. As shown in Fig. 4A and 4B, the peak current of Kv channels gradually decreased with repetitive application of pulses under both control conditions and in the presence of 10 µM clozapine. However, the decrease in Kv current was greater in the presence of clozapine. Indeed, the peak current of Kv channels was decreased by 11% and 18% under the control conditions, and by 18% and 24% in the presence of clozapine at 1 and 2 Hz, respectively. These results suggest that the clozapine-induced inhibition of Kv channels is use (state)-dependent.
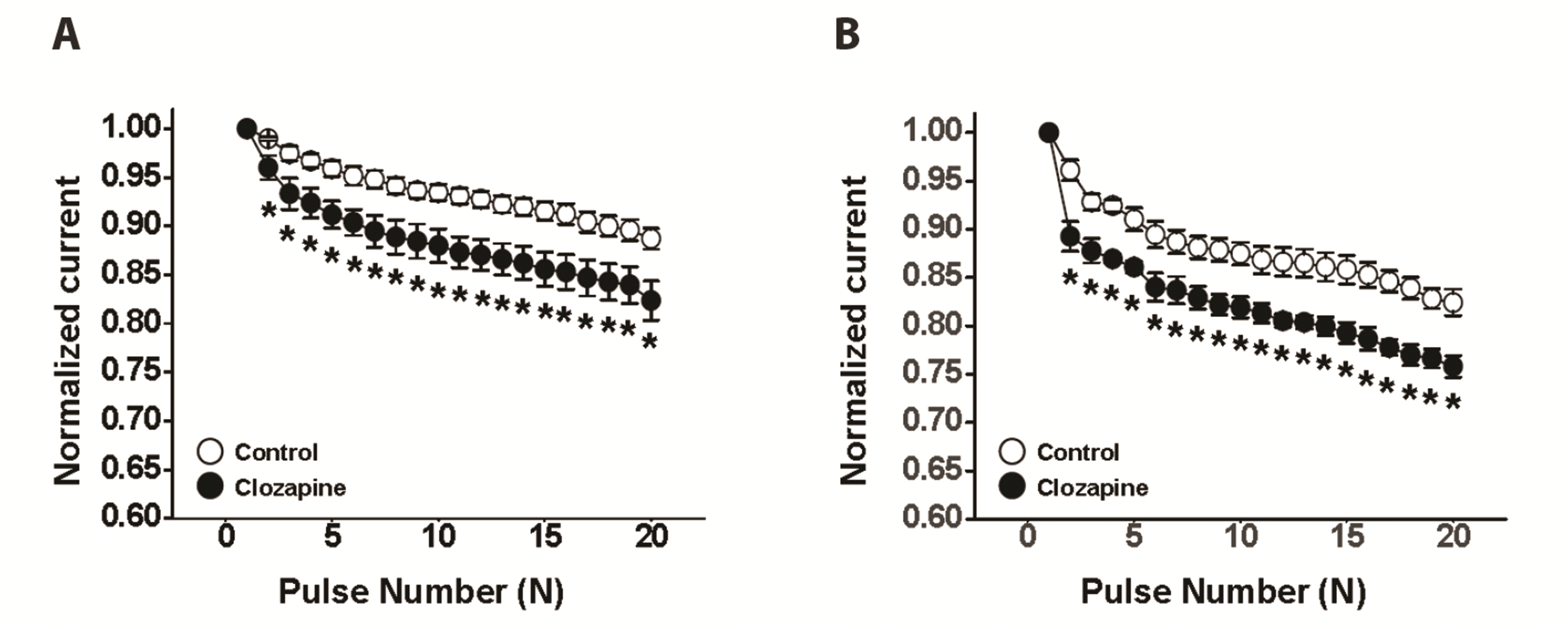 | Fig. 4Use (state)-dependent inhibitory effect of clozapine on voltage-dependent K+ (Kv) channels.Twenty repetitive depolarizing pulses from −80 to +60 mV with a duration of 150 ms were applied at frequencies of (A) 1 Hz and (B) 2 Hz, in the absence (○) and presence (●) of 10 μM clozapine. The peak currents of the Kv channels were normalized relative to the maximal peak current generated by the first pulse, and then plotted against the pulse numbers. n = 11. Data are mean ± SEM. *p < 0.05 (control vs. clozapine, at each pulse number).
|
Effects of clozapine on the recovery kinetics of Kv currents
To confirm the use (state)-dependent inhibition of Kv currents by clozapine, the time courses of recovery in the absence and presence of 10 µM clozapine were measured by applying a twin pulse protocol, as shown in Fig. 5 (inset). The interval between the pulses was gradually increased from 30 to 7,000 ms. The peak current of Kv channels at the second pulse was normalized by the peak current induced by the first pulse, and then plotted against the interpulse interval. The recovery time constants from inactivation in both the absence and presence of clozapine were fitted by a single exponential function with recovery time constants of 1,082.84 ± 238.79 and 1,996.94 ± 349.16, respectively (Fig. 5). The increased recovery time constant in the presence of clozapine explained the use (state)-dependent inhibition.
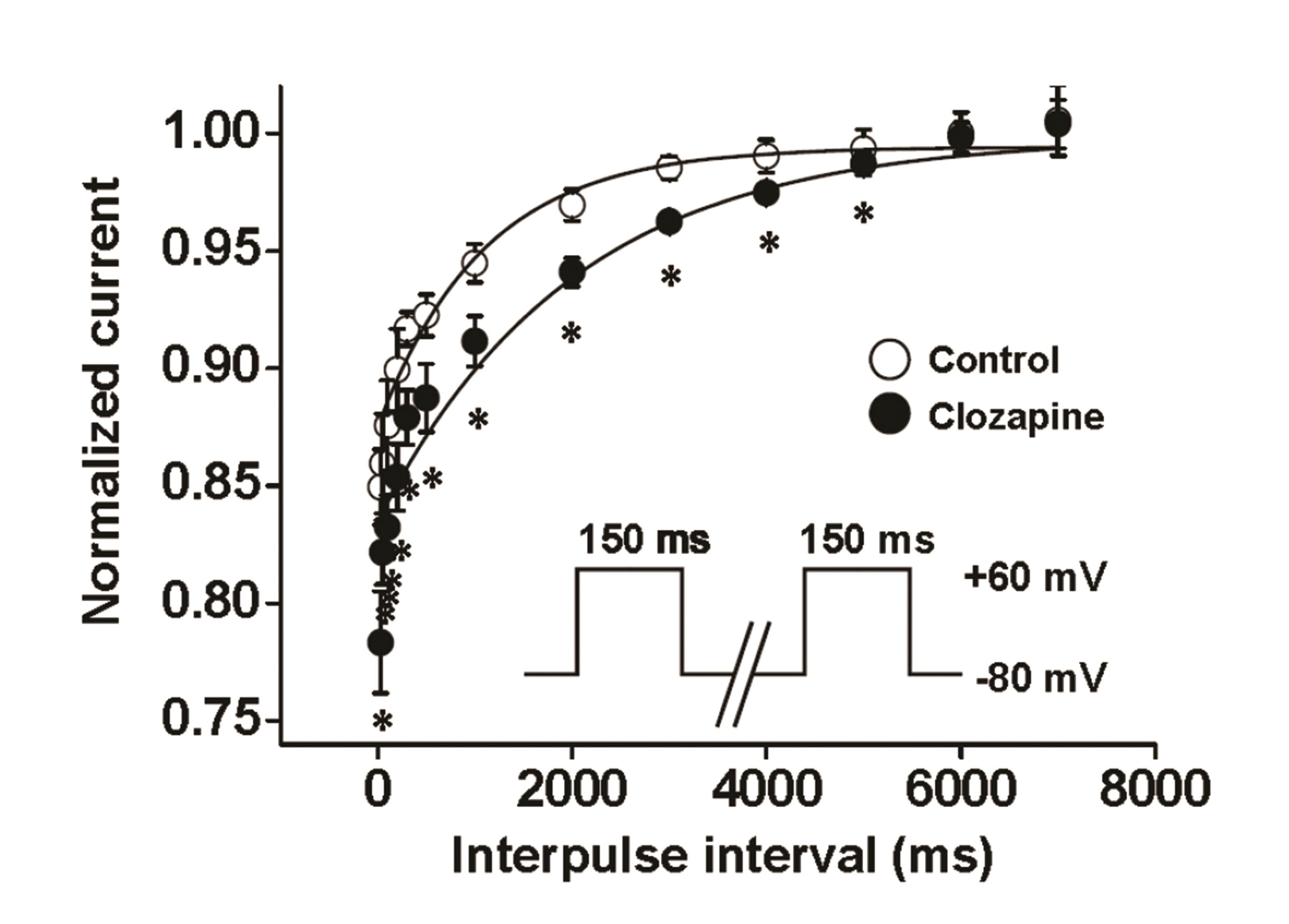 | Fig. 5Time course of recovery from clozapine-induced inhibition of voltage-dependent K+ (Kv) channels.The recovery time constants were measured by applying an identical twin pulse, as shown in the inset. A 150-ms depolarizing pre-pulse from −80 to +60 mV was followed by an identical second pulse with increasing intervals from 30 ms to 7 sec. Solid lines represent recovery of Kv currents in the absence (○) and presence (●) of 10 μM clozapine. n = 5. Data are mean ± SEM. *p < 0.05 (control vs. clozapine, at each time point).
|
Involvement of Kv subtypes on the inhibitory effect of clozapine on Kv channels
Vascular smooth muscles express several subtypes of Kv channels [17]. To determine the involvement of the Kv subtypes, we performed pretreatment with commercially available Kv subtype inhibitors: Kv1.5 subtype inhibitor (DPO-1, 1 µM), Kv2.1 subtype inhibitor (guangxitoxin, 30 nM), and Kv7 subtype inhibitor (linopirdine, 10 µM). The Kv currents were elicited by the same pulses depicted in Fig. 2. Pretreatment with 1 µM DPO-1 decreased the Kv current amplitudes, but additional application of 10 µM clozapine did not further inhibit the Kv current (Fig. 6A, B). Pretreatment with 30 nM guangxitoxin and 10 µM linopirdine also reduced the Kv currents (Fig. 6C, E). However, guangxitoxin and linopirdine partially blocked the inhibitory effect of clozapine (21% and 19%, respectively) compared to that induced by clozapine alone (45%) (Fig. 6D, F). These results suggest that Kv1.5 is the major subtype involved in clozapine-induced inhibition of Kv channels, and that Kv2.1 and Kv7 subtypes are partially involved in the inhibitory effects of clozapine on Kv currents.
 | Fig. 6Inhibitory effects of clozapine on voltage-dependent K+ (Kv)1.5, Kv2.1, and Kv7 subtypes.The representative Kv currents were elicited by applying 600-ms one-step depolarizing pulses of +60 mV from a holding potential of −80 mV. (A) Kv currents under control conditions, in the presence of DPO-1, and DPO-1 + clozapine. (B) Summary of the effects of DPO-1 on the inhibitory effect of clozapine on Kv currents. n = 5. NS, not significant (DPO-1 vs. DPO-1 + clozapine). (C) Kv currents under control conditions, in the presence of guangxitoxin, and in the presence of guangxitoxin + clozapine. (D) Summary of the effects of guangxitoxin on the inhibitory effect of clozapine on Kv currents. n = 4. *p < 0.05 (guangxitoxin vs. guangxitoxin + clozapine). (E) Kv currents under control conditions, in the presence of linopirdine, and in the presence of linopirdine + clozapine. (F) Summary of the effects of linopirdine on the inhibitory effect of clozapine on Kv currents. n = 5. *p < 0.05 (linopirdine vs. linopirdine + clozapine). Data are mean ± SEM.
|
Go to :
DISCUSSION
In the present study, we investigated the inhibitory effect of clozapine on Kv currents using native coronary arterial smooth muscle cells. The results suggest that clozapine inhibited Kv channels in a dose- and use (state)-dependent manner. These inhibitory effects of clozapine occurred regardless of the gating properties of the Kv channels. Furthermore, the major target of clozapine involved in Kv channel inhibition was the Kv1.5 subtype.
Our results suggest that the inhibition of Kv channels by clozapine is use (state)-dependent. As shown in Figs. 4 and 5, the reduction of Kv currents and the recovery time course from inactivation state were significantly increased in the presence of clozapine. These results suggest that clozapine inhibited the Kv current in a use (state)-dependent manner. However, clozapine did not change the steady-state activation or inactivation curve (Fig. 3), suggesting that clozapine inhibited Kv channels in a use (state)-dependent manner without affecting the gating properties. Use (state)-dependent channel inhibition can be divided into open- and inactivated-state inhibition. To determine whether clozapine-induced inhibition involves open- or inactivated-state inhibition, we analyzed the pattern of Kv current inhibition by clozapine. Generally, open-state channel inhibitors decrease the steady-state rather than peak of the Kv current. However, as shown in Figs. 1 and 2, clozapine inhibited Kv current amplitudes to the same degree at peak and steady-state, indicating that clozapine inhibits the channels primarily in the inactivated state. Furthermore, acceleration of current decay by clozapine, which is frequently observed with open-state channel inhibitors, was not observed in the present study. These observations suggest that clozapine-induced inhibition of Kv channels occurs in the inactivated state.
Based on our observations, the effect of clozapine seems not to be related to neurotransmitter secretion, but to direct inhibition of the Kv channels, for three main reasons. First, clozapine-induced inhibition of Kv currents was rapid. Indeed, the inhibition of Kv currents began as soon as clozapine was administered. Furthermore, the clozapine reaction reached a steady-state within 2 min. This instant reaction indicates that clozapine interacts directly with the Kv channels rather than through a neurotransmitter receptor-mediated signaling pathway. Second, we found that clozapine inhibited Kv channels in a use (state)-dependent manner (Figs. 4 and 5). The occurrence of inhibition in a specific state of a channel indicates that the inhibition occurs via a direct interaction with the channel or binding around the channel without antagonistic effects on neurotransmitter receptors. Third, neurotransmitter receptors are found in the central and peripheral nervous systems, and these receptors mediate both excitatory and inhibitory neurotransmission. However, our experimental system using single vascular smooth muscle cells excluded the involvement of neurotransmitter receptor-mediated responses. Based on these observations, we conclude that clozapine inhibited the Kv currents in a manner independent of neurotransmitter antagonism.
Kv channels in arterial smooth muscle are the most abundant channels in the cell membrane and play crucial roles in determining the resting membrane potential and vascular tone [10,11]. Additionally, alterations of Kv channel expression and/or function are closely associated with the onset of cardiovascular diseases including hypertension, diabetes, and atherosclerosis [13,14]. Therefore, it is important to identify the side effects of clinical drugs acting on arterial Kv channels, to prevent misinterpretation of vascular studies and the side effects involved in the clinical use of these drugs. Indeed, several recent studies have reported the side effects of atypical antipsychotics on arterial Kv channels independent of their antagonistic effects. Olanzapine, which acts on several neurotransmitter receptors, similar to clozapine, has been shown to inhibit vascular Kv channels with an IC50 value of 7.76 ± 1.80 µM [18]. However, olanzapine shifted the inactivation curve to a more negative potential and had no use (state) dependence. The other atypical antipsychotics, ziprasidone and iloperidone, also inhibited Kv currents with IC50 values of 0.39 ± 0.06 µM and 2.11 ± 0.5 µM, respectively [19,20]. They shifted the activation curve toward a more positive potential, and these effects on Kv channels were use (state)-dependent. Risperidone has also been shown to inhibit Kv currents with an IC50 value of 5.54 ± 0.66 µM [21]. In contrast to other atypical antipsychotics, risperidone shifted both activation and inactivation curves in a use (state)-dependent manner. Although we could not determine the precise reasons for these differences, they may have been due to the structural diversity among these drugs. Further research is needed to clarify this issue.
Kv channels consist of a number of diverse subfamilies (Kv1–Kv12). Five major families have been identified: Kv1, Kv2, Kv3, Kv4, and Kv7, and of these Kv1, Kv2, and Kv7 are specifically abundant in arterial smooth muscle [17,22]. Most studies of the expression and/or function of Kv subtypes have been conducted in mice, rats, and humans. Therefore, the previse Kv subtypes expressed in rabbit arteries are not known. However, recent studies suggested that Kv1.5, Kv2.1, and Kv7 subtypes are expressed in rabbit arteries [23-26], and inhibitors of these subtypes are commercially available. In fact, we found that inhibitors of these subtypes reduced the Kv currents (Fig. 6), suggesting that these subtypes are expressed in rabbit vascular smooth muscle cells. Therefore, we investigated their involvement in the effect of clozapine. Pretreatment with a Kv1.5 subtype inhibitor decreased the inhibitory effect of clozapine on Kv currents. Additionally, pretreatment with Kv2.1 or Kv7 subtype inhibitors partially reduced the inhibitory effect of clozapine on Kv currents. Based on these results, we conclude that the major subtype involved in clozapine-induced inhibition of Kv currents in rabbit coronary arterial smooth muscle cells is Kv1.5, and that Kv2.1 and Kv7 subtypes are partially involved. Previous studies have found that DPO-1 inhibited Kv1.5 channels with an IC50 value of 0.31 µM, and application of 1 µM DPO-1 completely inhibited Kv1.5 channels [27]. Guangxitoxin inhibited Kv2.1 channels with an IC50 value of 3 nM, and application of 100 nM guangxitoxin completely inhibited Kv2.1 channels [28]. Additionally, linopirdine inhibited Kv7 channels with an IC50 value of 2.4 µM, and 10 µM linopirdine sufficiently inhibited Kv7 channels [29]. Therefore, in our experiments, 1 µM DPO-1, 100 nM guangxitoxin, and 10 µM linopirdine could completely inhibit Kv1.5, Kv2.1, and Kv7 currents, respectively. However, it is possible that these have nonspecific effects on other Kv subtypes. Further research will be required to clarify the detailed involvement of Kv subtypes in the inhibitory effects of clozapine using specific Kv subtype expression systems.
Clozapine (Clozaril) is rapidly and completely absorbed into the gastrointestinal tract. The peak plasma concentration occurs within 1–4 h, and the Cmax value following administration of 100 mg is 1.72 ± 0.74 µM [30,31]. In the present study, clozapine decreased the Kv currents with an IC50 value of 7.84 ± 4.86 µM, which is approximately 4.5-fold higher than the peak plasma concentration. However, application at 1 µM decreased the Kv currents by 20% (Fig. 2B). Even small changes in K+ conductance can alter vascular tone and coronary blood flow, as vascular smooth muscle cells have high input resistance. Furthermore, overdose or abuse of clozapine could increase its peak plasma concentration. Therefore, clozapine should be prescribed with caution in patients with vascular diseases.
In conclusion, the present study demonstrated the inhibitory effects of clozapine on Kv channels in rabbit coronary arterial smooth muscle cells. These inhibitory effects occurred in a concentration- and use (state)-dependent manner without changing the gating properties of the channels. Additionally, the main target of clozapine was Kv1.5 subtype, with partial involvement of Kv2.1 and Kv7 subtypes.
Go to :
 XML Download
XML Download