

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The mitochondrial free radical theory suggests that mitochondrial oxidative injury is caused by unquenched increased reactive oxygen species (ROS) [1-3]. This is considered to be a key factor of age-related disease including neurodegenerative, cardiovascular, and malignant disease [2]. Mitochondria play a prominent role in regulating oxidant generation, cellular bioenergetics, and cell senescence and death [3]. During normal oxidative metabolism, mitochondrial ROS are neutralized using several matrix enzymes including Mn-superoxide dismutase 2, glutathione peroxidase 1 (GPx1), thioredoxin reductase 2 (Trx2). These enzymes act to provide a cellular reduction environment and to control the apoptotic pathway [4]. Additionally, normal mitochondrial oxidized/reduced nicotinamide dinucleotide ratio (NAD+/NADH) and reduced/oxidized glutathione ratio (GSH/GSSG) are essential components of a normal stable and balanced cellular redox state providing optimal metabolic activity [5]. However, oxidative stress sets in when there is excessive generation of ROS which disturb the normal equilibrium maintained between the oxidative and antioxidant systems. To this end, mitochondria are considered a major source of ROS and constitute a primary target of oxidative damage, thus lowering its efficiency and leading to more ROS generation and dysfunction. Disturbances in various mitochondrial enzyme activities and redox state have been shown to reflect cellular senescence [6,7].
Replicative senescence can be described as a cellular aging phenomenon that limits the number of cell divisions (doublings of in vitro cultured cells), leading to cell cycle arrest [6]. Such an arrest is triggered by many stress factors including oxidative stress, telomere shortening, oncogenic activation, therapeutic toxicity, and mitochondrial stress [8-10]. Senescent cells have been shown to accumulate in aged tissue [8,10], and to be associated with a variety of age-related disorders including pulmonary and renal fibrosis, atherosclerosis, hepatic steatosis, hypertension, and lung, breast, and colorectal cancer [11-15]. In addition, dysfunctional senescent cells have been associated with several mitochondrial defects including increased mass, respiratory chain defects, OS, mitochondrial DNA (mtDNA) mutations, and structural abnormalities [16].
Because of their pivotal role in cellular bioenergetics, physiological processes, and redox signaling, mitochondria are considered major contributors to senescence [17]. To this end, lowered mitochondrial respiratory chain activity, and its coupled oxidative phosphorylation, has been demonstrated using different types of model systems and senescent cells [18-20]. Furthermore, dysfunctional mitochondria exhibit elevated ROS which promotes DNA damage characteristic of senescent cells [21]. It is also noteworthy that mtDNA is significantly susceptible to ROS generation rates owing to its proximity to the production site. Subsequently, damaged mtDNA leads to impaired oxidative phosphorylation, which further stimulates ROS release [6]. Additionally, senescent cell mitochondria exhibit dysregulated metabolic activity adversely affecting the antioxidant capacity, TCA cycle, electron transport chain (ETC) activity, NAD+/NADH, NADP/NADPH, GSH/GSSG ratios, ATP levels, and important biological processes, most notably mitopathy, fusion, and fission [17,22,23].
Moreover, mitochondrial anabolic activity has been associated with cellular senescence [21,24]. Nevertheless, notwithstanding the rise in the mitochondrial pool, total ATP synthesis via oxidative phosphorylation and AMP-activated protein kinase has been shown to be significantly reduced during senescence [25]. Additionally, senescent cell mitochondria have reduced membrane potential and profound enzyme leakage [26]. In congruence, deviation of mitochondrial homeostasis supports the development and maintenance of senescence.
Although a very large number of publications have reported on mitochondrial, structural, metabolic, and molecular changes in senescent cells, such findings came in studies designed to investigate single specific features in different types of human tissues using different types of study models. In contrast, there has been few studies that describe a comprehensive metabolic profile in oxidatively stressed senescent cell mitochondria in the same type of human tissue. Furthermore, the theory of ROS being associated with senescence is still controversial. Hence, this work investigates major mitochondrial metabolic changes using senescent, oxidatively stressed human skin fibroblast cultures.
Go to : 
METHODS
Cell culture
Primary human neonatal dermal fibroblasts were purchased from Lonza Pharma Biotech (#CC-2509; Lonza Pharma Biotech, Basel, Switzerland). Cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) under standard growth conditions [27] and were free from Mycoplasma contamination. Cellular senescence was achieved by serial subculture (i.e., one population doubling) at passage 2 (P2) into P25 and P30.
Mitochondrial isolation
Mitochondria were isolated using Mitochondria/Cytosol Fractionation Kit (Abcam, Cambridge, MA, USA) according to the manufacturer’s protocol. Washed cells were suspended in cytosol extraction buffer and homogenized in an ice-cold Dounce tissue grinder. Homogenates were then centrifuged at 700 × g for 10 min at 4°C, and the supernatants were again centrifuged at 10,000 × g for 30 min at 4°C. The cytosolic fraction in the resultant supernatant was collected while the pellets were resuspended in mitochondrial extraction buffer and vortexed to prepare mitochondrial protein lysate. Both fractions were finally stored at –80°C. Table 1 shows that mitochondrial fractions are indeed enriched with specific enzymatic activities of cytochrome C oxidase (complex IV) and citrate synthase (CS) compared to cytosolic fractions.
Markers of cellular senescence
Phosphofructokinase (PFK) activity was assayed based on fructose1,6-diphosphate formation and monitoring NADH oxidation as per [27]. Lactate dehydrogenase (LDH) activity was derived from lactate formation and monitoring NADH oxidation as detailed in [27]. Glycogen phosphorylase (GP) was determined by the rate of glucose-1-phosphate formation and monitoring NADP reduction as detailed in [27]. Also, hydrogen peroxide (H2O2) generation was estimated by fluorescent substrate Amplex Red Reagent (Thermo Fisher, Waltham, MA, USA) at excitation and emission spectra of 530 and 590 nm, respectively [28], while superoxide anion (SA) radicals were estimated from blue formazan as per [27]. Moreover, malondialdehyde levels were used as a surrogate for lipid peroxidation (LPO) as per [29], whereas protein carbonylation content (PCC) was measured by reacting carbonylated proteins with dinitrophenylhydrazine as outlined by Reznick and Packer [30]. GSH and GSSG content was measured as previously described [31,32]. TrxR2 activity was measured using Cayman Chemical’s colorimetric assay kit (Ann Arbor, MI, USA) based on reduction of (5,5´-dithiobis[2-nitrobenzoic acid]). SOD2 activity was determined as per [33] whereas GPx1 and glutathione reductase (GR) were assayed in fibroblast sonicates based on GSH oxidation and reduction as previously described [34,35]. Additionally, the rate of protein synthesis (Table 2) was monitored using radiolabeled incorporation of L-[14C(U)]-Leucine as previously described [34].
Table 2
Time course incorporation of L-[14C(U)]-Leucine into protein in serially subcultured human fibroblasts (cpm/μg protein)
![]()
TCA cycle enzymes
CS activity was detected by monitoring the increase in absorbance at 413 nm was monitored for 10 min following the addition of 0.1 M oxaloacetate as previously shown by us [27]. The synthesis of aconitate from isocitrate in presence of 50 mM Tris-HCl (pH 7.4), 20 mM isocitrate, and 0.5 mM MnC12 was used to measure aconitase (ACN) activity by following the increase in absorbance at 240 nm according to Hausladen and Fridovich [36]. NAD+- and NADP+-isocitrate dehydrogenase (ICD) activities were measured using NAD+ and NADP+ reduction at 340 nm and 380 nm, as reported by Goncalves et al. [37]. The activity of α-ketoglutarate dehydrogenase (α-KGDH) was detected by the absorbance of NADH at 340 nm as described by Lai and Cooper [38]. The assay buffer consisted of 0.2 mM TPP, 2 mM NAD+, 0.2 mM CoA, 1 mM MgCl2, 0.3 mM DTT, 0.1% Triton X-100, 10 mM α-KG, 130 mM HEPES-Tris (pH 7.4). Succinate dehydrogenase (SDH) was assayed by the method of Tan et al. [39] in which succinate oxidation and concurrent dichloroindophenol reduction was monitored at 600 nm in presence of 0.25 M sucrose, 50 mM phosphate (pH 7.6), 60 μM 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinone (DB) or 2,3-dimethoxy-5-methyl-6-pentyl-1,4-benzoquinone (DPB), 1.0 mM potassium cyanide, and 1.0 mM EDTA. Malate dehydrogenase (MDH) activity was determined in a reaction mixture of 100 mM potassium phosphate buffer (pH 7.4) containing 10 μM rotenone, 0.15 mM NADH, and 0.33 mM oxaloacetate [40].
Nicotinamide cofactors
NAD+, NADH, NADP+, and NADPH were measured as we have previously reported [40]. Briefly, assay mixtures contained 50 mM glycylglycine, 0.5 mM EDTA (pH 7.4), 1 mM thiazolyl blue, 0.5 mM phenazine ethosulfate, and either 60 μg/ml alcohol dehydrogenase for NAD+ and NADH or 5 mM (3.5 U/ml) glucose-6-phosphate dehydrogenase (G6PD) for NADP+ and NADPH. The reaction was initiated by the addition of 0.6 M ethanol and the absorbance was monitored at 570 nm. Standards of NAD+ and NADP+ were accordingly used.
ETC enzymes
NADH-cytochrome C reductase (Complex I-III), succinate cytochrome C reductase (Complex II-III), and Complex IV activities were determined as previously reported by us [27]. In brief, Complex I-III activity was assayed by monitoring cytochrome C reduction at 550 nm, while Complex II-III activity was determined by the addition of 100 μM of cytochrome C and 10 mM of succinate and then following the reaction at 550 nm. Complex IV activity was detected by monitoring the decrease in absorbance at 550 nm as a function of cytochrome C oxidation.
ATP and ADP levels
ATP level was measured using a colormetric ATP Content Assay Kit (Solarbio Life Science, Beijing, China). Hexokinase hydrolyzes ATP into ADP during the formation of glucose-6-phosphate from glucose, then G6PD catalyzes the dehydrogenation of glucose-6-phosphate to form 6 phospho-gluconate with the concurrent reduction of NADP+ into NADPH, whose formation, detected at 340 nm, is used to measure ATP content [41]. ADP is converted to ATP and pyruvate which is measured at 570 nm and is proportional to ADP content [42].
Quantitative real-time PCR (qRT-PCR)
Gene expression of TrxR1, SOD2, GPX1, and GR was estimated using specific primers following RNA isolation with Solarbio’s Total RNA Extraction Kit and cDNA synthesis with Solarbio’s Maxima First Strand cDNA Synthesis Kit. Relative expression as fold change was calculated by the 2–ΔΔCT method using GAPDH as a reference gene [43].
Statistical analysis
Results are expressed as means ± SEM of duplicate measurements obtained from three independent experiments (n = 6). Data were analyzed with GraphPad Prism v9.2.0 (GraphPad Software, Inc., San Diego, CA, USA) using one-way ANOVA followed by Bonferroni’s post-hoc test, and statistical significance was defined by a p-value of < 0.05.
Go to :
RESULTS
Senescence markers are increased in serially subcultured fibroblasts
In order to biochemically identify senescent cells, glucose and glycogen catabolic enzyme activities were determined in P5 through P35 cells. As shown in Fig. 1A–C, PFK, LDH, and GP underwent progressive increases in activity attaining statistical significance at P20 onward. PFK activity increased from control values of 19.50 ± 0.76 at P5 to 42.17 ± 1.42 μM/min/mg protein at P20, peaking at P35 with values of 79.83 ± 1.53 μM/min/mg protein (Fig. 1A). Similarly, LDH exhibited a gradual, significant increase from 155 ± 7.63 in control cells to 396.7 ± 14.70 μM/min/mg protein at P20, reaching a maximum activity at P35 with values of 504.2 ± 11.43 μM/min/mg protein (Fig. 1B). As for GP, activities amounted to 98.83 ± 3.53 in P5 control cells, and significantly increased at P20 to 414.0 ± 11.11 μM/min/mg protein at P20 with a peak activity of 510.5 ± 11.06 μM/min/mg protein at P35 (Fig. 1C).
 | Fig. 1Identification of senescent fibroblasts by monitoring total cellular glucose and glycogen degradative enzyme activities and oxidative stress markers in confluent serial subcultures.Enzyme activities of (A) phosphofructokinase (PFK), (B) lactate dehydrogenase (LDH), and (C) glycogen phosphorylase (GP), along with levels of (D) hydrogen peroxide (H2O2) and (E) superoxide anions (SA), (F) lipid peroxides (LPO), (G) protein carbonyl content (PCC), and (H) oxidized glutathione (GSSG) in P5-P35 confluent cells. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 indicate significant difference from control P5 values (n = 6).
|
Senescent cells exhibit profound oxidative stress
To assess the oxidative stress status of senescent cells, and to also identify the latter, major indicators of oxidative stress were examined. As shown in Fig. 1D, a significant increase in the generation rates of H2O2, compared to P5 values of 1.70 ± 0.02 μM/min/mg protein, were observed at P20 (4.25 ± 0.05 μM/min/mg protein), reaching a maximum of 6.65 ± 0.45 μM/min/mg protein at P35.
Likewise, the baseline level of SA generation was 2.63 ± 0.18 μM/min/mg protein at P5, which significantly increased at P20 to 4.65 ± 0.45 and peaked at P35 to 10.74 ± 0.38 μM/min/mg protein (Fig. 1E). Moreover, LPO at P5 was 40.15 ± 3.25 μM/min/mg protein which was significantly elevated at P20 to 60.80 ± 2.40 μM/min/mg protein with a maximum of 87.30 ± 5.0 μM/min/mg protein at P35 (Fig. 1F). PCC also showed a similar pattern with P5 values of 5.39 ± 0.18 nM/min/mg protein reaching statistical significance at P20 to 11.69 ± 1.04 nM/min/mg protein and peaking at P35 to 26.51 ± 2.03 nM/min/mg protein (Fig. 1G). Finally, as depicted in Fig. 1H accumulation of GSSG was also significantly increased from P5 levels of 0.85 ± 0.07 nM/min/mg protein to 1.77 ± 0.11 and to 3.60 ± 0.20 nM/min/mg protein at P20 and P35, respectively.
Based on the above data and in the current study, P5 subcultures have been regarded as primary (young controls), whereas those at P20 as early senescent and those at P25 through P35 as senescent.
Senescence disrupts mitochondrial TCA cycle enzyme activities
In an attempt to probe potential alterations in mitochondrial function associated with senescence, key TCA cycle enzyme activities were assayed in mitochondrial sonicates of serially subcultured cells.
As demonstrated in Fig. 2A, control ACN activity in P5 cells (8.33 ± 0.54 μM/min/mg protein) was severely diminished in P25 and P35 cells to 1.73 ± 0.42 and 1.61 ± 0.32 μM/min/mg protein, respectively. Fig. 2B shows a significant decrease in the activity of α-KGDH from 2.18 ± 0.16 in P5 cells to 0.58 ± 0.10 and to 0.51 ± 0.10 μM/min/mg protein in P25 and P35 cells, respectively. In Fig. 2C, baseline SDH activities averaging of 3.70 ± 0.08 μM/min/mg protein were also significantly inhibited to 0.95 ± 0.08 μM/min/mg protein at P25 and 0.84 ± 0.06 μM/min/mg protein at P35. This pattern was also reciprocated with MDH activity, as depicted in Fig. 2D, which has significantly fallen from P5 values of 1.49 ± 0.11 μM/min/mg protein to 0.33 ± 0.01 and 0.29 ± 0.18 μM/min/mg protein in P25 and P35, respectively.
 | Fig. 2Mitochondrial specific activities of TCA cycle enzymes in primary P5 and senescent P25 and P35 fibroblasts.Enzyme activities of (A) aconitase (can), (B) α-ketoglutarate dehydrogenase (α-KGDH), (C) succinate dehydrogenase (SDH), (D) malate dehydrogenase (MDH), (E) NAD+- isocitrate dehydrogenase (ICD), and (F) NADP+-ICD in young (P5) and senescent (P25 and P35) confluent cells. ***p < 0.001 and ****p < 0.0001 indicate significant difference from control P5 values (n = 6).
|
Furthermore, Fig. 2E shows that the two forms of ICD, NAD+-ICD and NADP+-ICD, had control activities of 1.00 ± 0.05 and 5.35 ± 0.27 μM/min/mg protein which underwent significant decrease in P25 cells to 0.29 ± 0.01 and 1.75 ± 0.04 μM/min/mg protein and in P35 cells to 0.26 ± 0.01 and 1.41 ± 0.05 μM/min/mg protein, respectively.
Senescence is associated with reduced ETC enzyme activities
The potential influence of cellular senescence on ETC enzymes in mitochondrial sonicates was also studied. Fig. 3A shows that the activity of Complex I–III was significantly reduced in P25 cells (46.17 ± 1.12 nM/min/mg protein) and in P35 cells (42.73 ± 2.24 nM/min/mg protein) compared to P5 cells (83.29 ± 1.40 nM/min/mg protein). The activity of Complex II-III exhibited a similar pattern, showing baseline values of 97.77 ± 6.28 nM/min/mg protein in P5 cells, which was significantly decreased at P25 to 45.01 ± 2.33 nM/min/mg protein and at P35 to 45.62 ± 1.27 nM/min/mg protein (Fig. 3B). Complex IV similarly showed significant inhibition from 43.70 ± 1.57 nM/min/mg protein (P5) to 23.23 ± 1.35 nM/min/mg protein (P25) and to 21.05 ± 1.30 nM/min/mg protein (P35) as demonstrated in Fig. 3C. Finally, Fig. 3D shows that CS activity; an indicator of mitochondrial function, also decreased from 71.74 ± 1.44 nM/min/mg protein (P5) to 25.38 ± 2.27 nM/min/mg protein (P25) and 18.41 ± 0.52 nM/min/mg protein (P35).
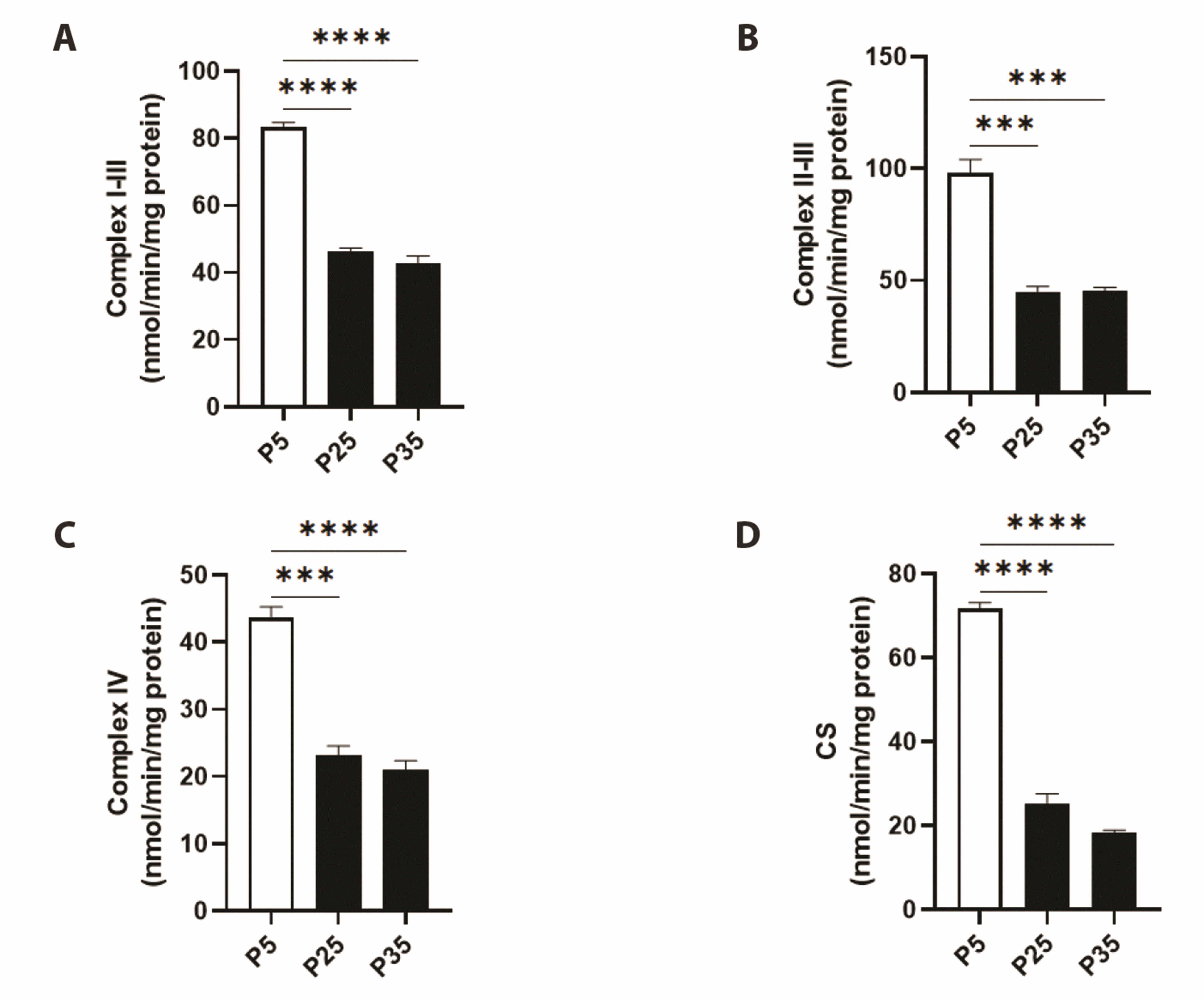 | Fig. 3Mitochondrial specific activities of electron transport chain (ETC) enzymes in primary P5 and senescent P25 and P35 fibroblasts.Enzyme activities of (A) complex I-III, (B) complex II-III, (C) complex IV, and (D) citrate synthase (CS) in young (P5) and senescent (P25 and P35) confluent cells. ***p < 0.001 and ****p < 0.0001 indicate significant difference from control P5 values (n = 6).
|
Senescence promotes mitochondrial oxidative stress
The redox state of senescent mitochondria was assessed by measuring the generation rate of H2O2, SA, LPO, and PCC. Fig. 4A shows that baseline levels of H2O2 in P5 mitochondria were 5.90 ± 0.40 μM/min/mg protein which were significant increased at P25 to 18.73 ± 0.38 μM/min/mg protein and at P35 to 20.90 ± 0.67 μM/min/mg protein. Fig. 4B depicts significant elevation of SA levels from 8.51 ± 0.74 μM/min/mg protein in P5 mitochondria to 30.15 ± 0.82 μM/min/mg protein and to 33.63 ± 0.53 μM/min/mg protein in P25 and P35 mitochondria, respectively.
 | Fig. 4Mitochondrial oxidative stress markers in primary P5 and senescent P25 and P35 fibroblasts.Generation levels of (A) hydrogen peroxide (H2O2), (B) superoxide anions (SA), (C) lipid peroxides (LPO), and (D) protein carbonyl content (PCC), and transcriptional and enzyme activities of thioredoxin reductase 2 (TrxR2) (E, F) and superoxide dismutase 2 (SOD2) (G, H) in young (P5) and senescent (P25 and P35) confluent cells. **p < 0.01), ***p < 0.001, and ****p < 0.0001 indicate significant difference from control P5 values (n = 6).
|
Likewise, in Fig. 4C, generation of LPO significantly increased from 151.1 ± 6.93 μM/min/mg protein (P5) to 462.3 ± 16.78 μM/min/mg protein (P25) and to 494.3 ± 19.67 μM/min/mg protein (P35). Carbonylated proteins were also significantly increased at P25 to 54.64 ± 2.42 nM/min/mg protein and at P35 to 59.57 ± 3.87 nM/min/mg protein, compared to 17.41 ± 0.67 nM/min/mg protein at P5 (Fig. 4D).
Next, we determined the transcriptional and enzymatic activities of TrxR2 and SOD2. As shown in Fig. 4E, TrxR2 expression significantly increased by 1.85 ± 0.06 and 1.81 ± 0.08 folds in P25 and P35, respectively, compared to P5 cells. Similarly, TrxR2 showed a significant increase in activity from 14.07 ± 0.29 nM/min/mg protein in P5 mitochondria to 21.01 ± 0.95 nM/min/mg protein and to 23.10 ± 0.73 nM/min/mg protein in P25 and P35 mitochondria (Fig. 4F). SOD2 expression was also elevated by 1.87 ± 0.13 folds in P25 and by 1.92 ± 0.13 folds in P35 mitochondria as compared to P5 (Fig. 4G). Significant elevations in SOD2 activity were also recorded, as seen in Fig. 4H, as P5 mitochondria showed an average of 31.49 ± 1.30 μM/min/mg protein whereas those of P25 had 46.41 ± 1.20 μM/min/mg protein and those of P35 had 49.91 ± 0.79 μM/min/mg protein.
To assess disruptions in glutathione metabolism, transcriptional and enzymatic activities of GPx1 and GR, along with GSH and GSSG levels were examined. Fig. 5A shows that mRNA expression of GPx1 increased by 1.85 ± 0.06 folds (P25) and 1.98 ± 0.12 folds (P35) compared to P5, as did GR expression (Fig. 5B) which increased by 1.68 ± 0.07 and 1.96 ± 0.08 folds in P25 and P35, respectively. In Fig. 5C, GPx1 activity at P5 had values of 2.96 ± 0.23 μM/min/mg protein, which significantly increased to 5.01 ± 0.10 μM/min/mg protein (P25) and to 5.04 ± 0.20 μM/min/mg protein (P35). Consistent with gene expression, GR enzymatic activity (Fig. 5D) was also significantly elevated from baseline P5 values of 3.10 ± 0.03 μM/min/mg protein to 4.02 ± 0.14 μM/min/mg protein (P25) and to 4.45 ± 0.25 μM/min/mg protein (P35). In Fig. 5E, GSH at P5 equaled 35.23 ± 1.27 nM/min/mg protein which was significantly reduced to 20.62 ± 1.25 nM/min/mg protein at P25 and to 19.91 ± 1.62 nM/min/mg protein at P35. Conversely, in Fig. 5F, GSSG at P5 was 0.55 ± 0.08 nM/min/mg protein with significant accumulation at P25 equaling 1.75 ± 0.06 nM/min/mg protein and at P35 equaling 1.85 ± 0.05 nM/min/mg protein. Accordingly, the GSH/GSSG ratio was severely diminished at P25 reaching 11.79 ± 1.00 and at P35 with 10.81 ± 1.18, in comparison to 66.30 ± 9.52 at P5 (Fig. 5G).
 | Fig. 5Mitochondrial glutathione status in primary P5 and senescent P25 and P35 fibroblasts.Transcriptional activities of (A) glutathione peroxidase 1 (GPx1) and (B) glutathione reductase (GR), and enzyme activities of (C) GPx1 and (D) GR, along with levels of (E) reduced glutathione (GSH), (F) oxidized glutathione (GSSG), and (G) GSH/GSSG ratio in young (P5) and senescent (P25 and P35) confluent cells. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 indicate significant difference from control P5 values (n = 6).
|
Nicotinamide cofactors are disrupted in senescent mitochondria
To investigate the influence of senescence on nicotinamide cofactors, the levels of mitochondrial NAD+, NADH, NADP+, NADPH were assayed. Fig. 6A shows that NAD+ was significantly decreased from 272.0 ± 4.90 pM/min/mg protein at P5 to 204.3 ± 2.96 pM/min/mg protein at P25 and to 199.7 ± 3.18 pM/min/mg protein at P35. This was accompanied by a concurrent reduction in NADH, as seen in Fig. 6B, from 33.24 ± 0.57 pM/min/mg protein to 13.19 ± 0.33 pM/min/mg protein at P25 and to 12.46 ± 0.27 pM/min/mg protein at P35. This resulted in a significant increase in NAD+/NADH ratio from 8.18 ± 0.13 at P5 to 15.52 ± 0.62 at P25 and to 16.04 ± 0.37 at P35 (Fig. 6C).
 | Fig. 6Mitochondrial NAD+, NADH, NADP+, and NADPH levels in primary P5 and senescent P25 and P35 fibroblasts.Levels of (A) NAD+, (B) NADH, (C) NAD+/NADH ratio, (D) NADP+, (E) NADPH, and (F) NADP+/NADPH ratio in young (P5) and senescent (P25 and P35) confluent cells. **p < 0.01, ***p < 0.001, and ****p < 0.0001 indicate significant difference from control P5 values (n = 6).
|
Similarly, in Fig. 6D, NADP+ levels at P5 were 34.30 ± 0.46 pM/min/mg protein which significantly decreased to 26.83 ± 0.64 pM/min/mg protein at P25 and to 25.84 ± 1.11 pM/min/mg protein at P35. NADPH levels were also significantly reduced as seen in Fig. 6E from 81.0 ± 0.57 pM/min/mg protein at P5 to 34.43 ± 0.50 pM/min/mg protein at P25 and to 31.57 ± 0.73 pM/min/mg protein at P35. As a result, the ratio of NADP+ to NADPH was significantly increased to 0.77 ± 0.01 at P25 and to 0.82 ± 0.05 at P35 compared to 0.42 ± 0.01 at P5 (Fig. 6F).
Senescence promotes metabolic exhaustion
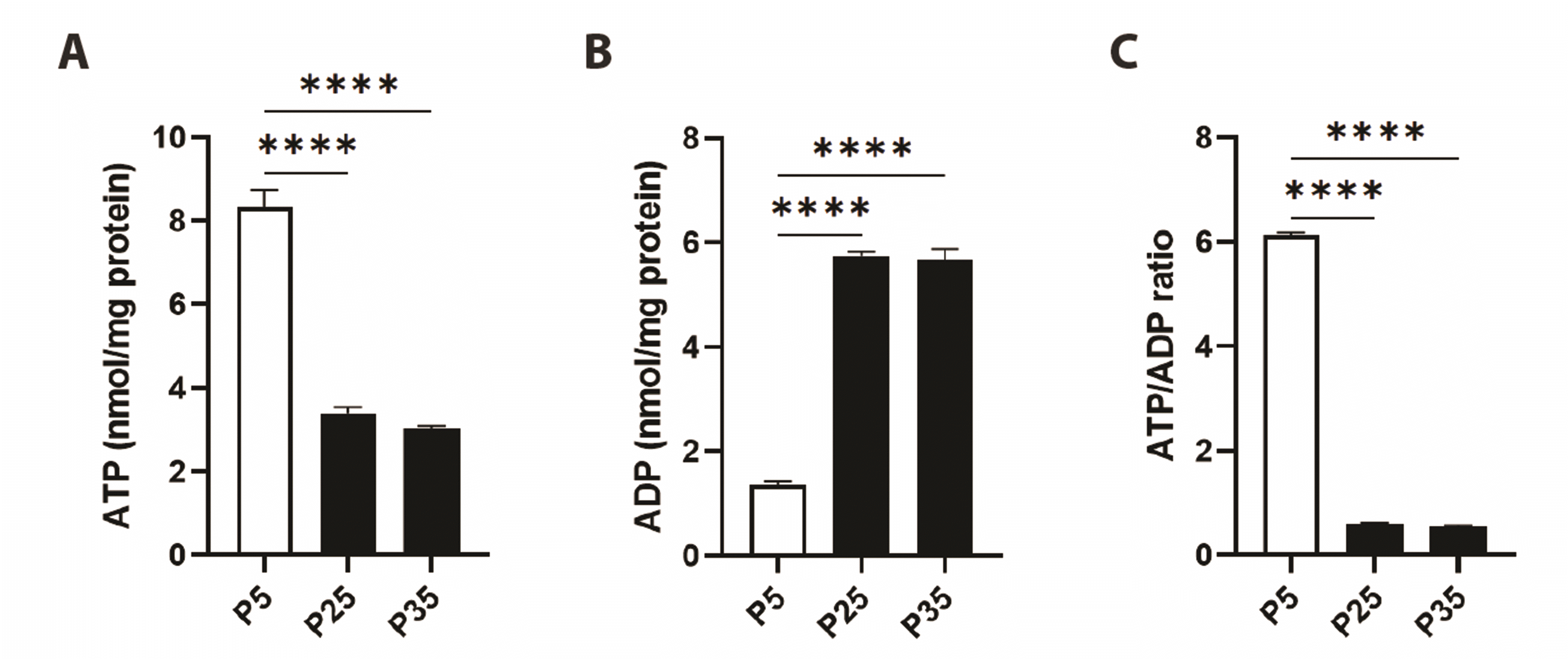
In order to evaluate whether senescence is associated with energy depletion, we measured ATP and ADP levels in mitochondrial sonicates of young and senescent cells. As seen in Fig. 7A, P5 cells showed ATP levels of 8.32 ± 0.42 nM/min/mg protein which was significantly reduced at P25 to 3.38 ± 0.15 nM/min/mg protein and to 3.04 ± 0.05 nM/min/mg protein at P35. In Fig. 7B, a concurrent increase in ADP levels is observed from 1.35 ± 0.06 nM/min/mg protein at P5 to 5.73 ± 0.09 nM/min/mg protein at P25 and to 5.66 ± 0.22 nM/min/mg protein at P35. Accordingly, the ATP/ADP ratio was significantly reduced from 6.13 ± 0.04 at P5 to 0.59 ± 0.03 at P25 and to 0.53 ± 0.03 at P35 (Fig. 7C).
Go to :
DISCUSSION
Cultured human foreskin fibroblasts offer a successful model which has been extensively used in our laboratory to study intracellular metabolic changes associated with the decline of the antioxidant capacity with age, venom toxicity and its effect on the molecular, and metabolic activity of tissue and the status of oxidative stress markers in placental tissue of patients with several types of reproductive disease [27,29,31-34,44,45]. Fibroblasts exhibit rich metabolic activity, and the results obtained can be used to understand underlying mechanisms in other tissues. Fetal bovine serum added to the medium provided protein spreading factors for the cells, and the metal cations Se4+, Zn2+, Cu2+, and Mn2+ at normal human plasma levels. Such concentrations are optimal and essential to ensure maximal specific activities of the investigated mitochondrial antioxidant enzymes. These trace metalloid elements act as cofactors for the enzymes and are able to modulate gene expression of the investigated antioxidant enzymes and enhance proliferation of subcultures at senescence, thereby offering a protection against oxidative stress [28,44,46].
In the present study, care was taken to identify the culture passage at which senescence begins and continues. We have previously demonstrated that P5 and P10-cultured cells need 24 hours to reach confluence and maximal rates of radiolabeled leucine and thymidine incorporation into protein and DNA. Additionally, P15, P20, P25, P30, and P35 cultures required 48, 96 and 120 hours respectively. Hence, cells at different passages were harvested at such times post-culture. In the current study, we used the same harvesting times obtained by us when using human skin fibroblasts for various investigations [31,34,40]. Application of these harvesting times ensured that the protein and DNA yields of all subcultures could not be affected by senescence, and that changes in any of the investigated parameters are attributed to senescence.
Key glucose and glycogen degradative enzyme activities (PFK, LDH, and GP) underwent significant increase beginning in fibroblasts at P20, and gradually become much higher in magnitude at P25, P30, and P35 (Fig. 1A–C). Such results were in very close agreement with previous findings [34,47,48]. This suggested that the enhanced glycolytic metabolism is caused by senescence, and not related to hypoxia since cells were cultured in an atmosphere contenting 18% oxygen. Moreover, the cellular generation rates of all the major oxidative stress markers investigated (H2O2, SA, LPO, PCC, and GSSG) peaked in value in P30 and P35 cells compared against P5 cells (Fig. 1D–H).
The replicative senescence of cultured cells has been shown to reflect many aspects of organismal aging, most notably the increase in the number of senescent cells with aging [49-51]. As previously reported, replicative senescence of serially culture fibroblasts has been linked with a number of biochemical and morphological changes. These involve accumulation of oxidized protein, telomere shortening, flattened cellular morphology, cell enlargement, reduced replication and growth rates, enhanced glucose and glycogen degradative enzyme activities, increased generation of oxidative stress markers and an increase in the cells number showing β-galactosidase activity [29,31,33,34,50-53]. Moreover, such changes have been associated with a decrease in antioxidant defense mechanisms [52].
The presently reported specific activities of all investigated TCA enzymes were similar in range to those previously reported [40]. As indicated in Fig. 2, mitochondrial activities of ACN, SDH, MDH, NAD+-ICD plus none-TCA NADP+-ICD underwent highly significant decreases in senescent P25 and P35 cells compared to the those recorded for P5 cells. Such reductions were also similar in magnitude when expressed in terms of mitochondrial DNA rather than protein (data not shown). This lowered rate of TCA cycle could have been due to decreased glucose uptake by the senescent fibroblasts. However, this is highly unlikely since the activities of key glucose and glycogen degradative enzymes were markedly increased in senescent P25 and P35 cells (Fig. 1A–C). Thus, it is likely that the biochemical features of senescence such as oxidative stress may be one of the causes and is directed at the mitochondrial or cellular enzyme gene expression level, a phenomenon that needs further investigation. The above findings suggest that mitochondria of senescent cells produce less energy and are dysfunctional.
In congruence with the decline in TCA cycle activity, investigated activities of complex I–III, complex II–III, complex IV, and the mitochondrial marker CS exhibited very significant reduction in senescent P25 and P35 cells compared against those recorded for P5 cells (Fig. 3). The noted specific activities of all the investigated respiratory chain enzymes and CS obtained for P5 primary cells were within normal range, and very similar to those reported in the literature by us and others [27,54,55]. Accordingly, mitochondria of P5 cells were normally functional, and the lowered enzyme activities were due to senescence. Our results are in agreement to those reported previously in aged culture human fibroblasts [54], and mitochondria isolated from skeletal muscles of aged rats and humans [56].
Despite the low activities of all the mitochondrial respiratory chain enzymes, the protein content of the senescent P25 and P30 cells was not affected. Such results support the possibility of an increased number of dysfunctional mitochondria. To this end, electron micrographs of senescent P30 fibroblasts are required to examine any mitochondrial ultrastructure variations. The decrease in CS activity suggests that senescent cells are undergoing a lowered rate of mitochondrial oxidative metabolism. Considering that it approximately takes 2–3 weeks to reach P25 from P15, our results seem to suggest that the reduction in TCA cycle and respiratory chain activities is a time-dependent genetic program. As a consequence of the decline in the respiratory chain activity, the amount of energy released from electron transport will have been reduced.
The GPx1/GR antioxidant system is a ROS quencher that prevents cellular LPO and depletion of thiols, and maintains intramitochondrial and intracellular homoeostasis and redox balance [57]. This redox homoeostasis is primarily substantiated by GSH. GSH acts as a natural non-enzymatic antioxidant and a potential reducing agent, and is reported to protect the body system against the destructive effects of oxidative stress [58]. GSH depletion (Fig. 5E–G) leads to accumulation of SA (Figs. 1E and 4B) which plays a toxic role but cells have the ability to recycle GSH via the activity of the redox cycle. In this pathway, GSH is used as a co-substrate by GPx1 to decompose H2O2 (Figs. 1D and 4A) as well as LPO (Figs. 1F and 4C) [59], resulting in their reduction to water and production of GSSG. The latter is then reduced back to GSH by GR, via a reaction that requires NADPH generated by glucose-6-phosphate dehydrogenase. Consequently, the GSH/GSSG ratio normally maintained at a high value, represent the major cellular and intracompartmental redox buffer, and serve as an index of the cellular redox environment [60].
Results of the present study showed that mitochondria of senescent P25 and P35 cells exhibited dramatic increases in the level of GSSG (Fig. 5F, G). The above noted GSH decreases could have been a result of GSH reacting directly with excessive mitochondrial amounts of H2O2 and SA generated by the senescent cells, thus giving rise to increased GSSG levels. GR mitochondrial activity has also undergone slight increases (Fig. 5D), and this could have been another reason for the noted drop in mitochondrial GSH. GSH levels are also controlled by its de novo synthesis catalyzed by ƴ-glutamate cystine ligase and GR [61], which could have dropped in activity. Mitochondrial gene expression levels of these enzymes need to be examined in P25 and P35 senescent cells. Collectively, the current results indicate that the normally balanced redox state seen in mitochondria of primary P5 cells has shifted in P25 and P35 senescent cells towards oxidative metabolism and oxidative stress.
The NAD+/NADH ratio plays a significant role in regulating the intramitochondrial redox state and provides a major source of electrons for the metabolic activity of many enzymes. However, a high NAD+/NADH ratio indicates that it is sensitive to alterations in NADH concentration. Fig. 6A indicates that senescent P25 and P30 cells underwent a significant drop in NAD+ levels. This could have also contributed to the decreased dehydrogenase activities although it was of a lower magnitude than that noted in NADH levels (Fig. 6B), thereby causing a total increase in the NAD+/NADH ratio (Fig. 6C).
The drop in mitochondrial NAD+ concentrations could have been caused by the observed surge in oxidative stress of P25 and P30 cells mitochondria, a phenomenon that warrants further examination. Such an investigation should examine the activities of various key NAD+ synthesis enzymes including nicotinamide/nicotinate mononucleotide adenylyltransferase, nicotinamide phosphoribosyl- transferase and NAD+ synthetase.
The phosphorylated form of oxidized nicotinamide adenine dinucleotide (NADP+) is synthesized intracellularly by the enzyme NAD+ kinase. It is mainly present in the reduced form (NADPH) which acts as a reducing agent for enhancement of redox reactions linked with anabolic pathways. The NADP+/NADPH ratio has a low value when normal conditions prevail in mitochondria. Results of the current study clearly show that NADP+ and NADPH levels in mitochondria of senescent P25 and P30 cells underwent significant reductions when compared to those documented for P5 primary cells (Fig. 6D–F). Accordingly, NADP+-ICD activity was also significantly diminished (Fig. 2F). Since the drop in NADP+/NADPH ratio (Fig. 6F) was of similar magnitude with that observed for the NAD+/NADH ratio (Fig. 6C), it is envisaged that the mitochondrial oxidative stress linked with senescent P25 and P35 cells occurs as a result of the same mechanism. Hence, there is a need to investigate the NAD+-kinase activity in senescent cells.
Results presented in Fig. 7A show that the amount of ATP synthesized in mitochondria of passage 25 and passage 35 cells was very significantly lowered when compared against that recorded for primary P5 cells. In contrast, the level of ADP in mitochondria of P25 and P30 cells was highly increased (Fig. 7B). These results lead to a very significantly lowered ATP/ADP ratios in P25 and P30 cells compared against the ratios recorded for P5 cells (Fig. 7C). This profound energy depletion is compatible with the impaired antioxidant capacity observed throughout the study.
In conclusion, the current study presents detailed mechanistic insights into the oxidative injury accompanying cellular senescence using an in vitro model system (Fig. 8). Importantly, oxidative stress has been identified as a major contributor to many life-threatening conditions associated with aging, most notably malignancy [62], arthritis [63], and cardiovascular and neurodegenerative disease [64]. Antioxidants may, therefore, potentially be invaluable prophylactic or preventive tools to reduce the global burden of oxidative injury characteristic of age-related disease.
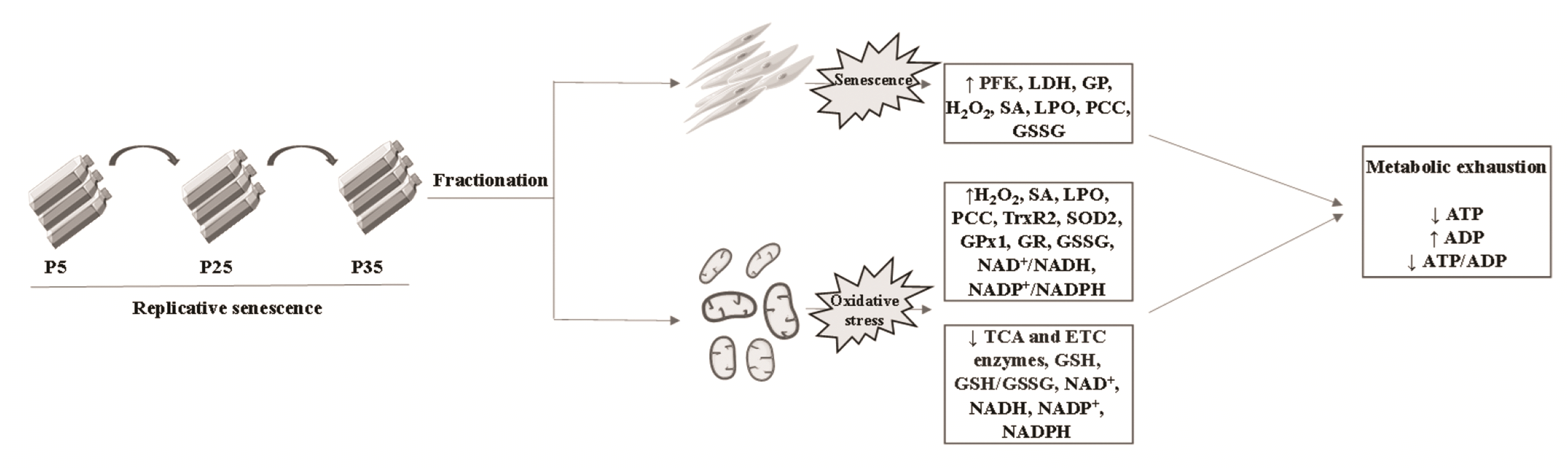 | Fig. 8A working model of mitochondrial senescence [65].Replicative senescence induces metabolic changes in mitochondria evident as accumulation of ROS, shutdown of TCA and ETC pathways, glutathione depletion, and energy exhaustion. ROS, reactive oxygen species; ETC, electron transport chain; PFK, phosphofructokinase; LDH, lactate dehydrogenase; GP, glycogen phosphorylase; H2O2, hydrogen peroxide; SA, superoxide anions; LPO, lipid peroxides; PCC, protein carbonyl content; GSSG, oxidized glutathione; TrxR2, thioredoxin reductase 2; SOD2, superoxide dismutase 2; GPx1, glutathione peroxidase 1; GR, glutathione reductase; GSH, reduced glutathione.
|
Go to :
 XML Download
XML Download