

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Hereditary hemolytic anemia (HHA) is a rare but highly heterogeneous inherited genetic disease characterized by anemia due to premature red blood cell (RBC) destruction and intrinsic RBC defects [1]. The three main etiologies of HHA are disorders of the RBC membrane (membranopathy), disorders of the RBC enzyme (enzymopathy), and disorders of hemoglobin (hemoglobinopathy) [1]. The prevalence of HHA is very low in Korea because hereditary spherocytosis (HS) is less common in Asians than in Caucasians, occurring in 1 per 5,000 population, and Korea does not belong to the thalassemia belt [2]. However, the incidence of hemoglobinopathy in Korea has increased in recent years due to an increase in immigrants, especially women, from Southeast Asia through international marriages [3, 4]. The awareness of clinicians about the disease and advances in the diagnostic technology also may have influenced the epidemiology of HHA in Korea [5, 6].
The RBC Disorder Working Party (WP), previously named HHA WP, of the Korean Society of Hematology (KSH), conducted a 10-year nationwide multicenter retrospective epidemiologic study of HHA in Korea from 2007 to 2016 [5]. Compared to the previous 10-year nationwide epidemiologic study of HHA in Korea from 1997 to 2006 [7], the proportions of hemoglobinopathies and enzymopathies have increased significantly in the last 10 years. Among the 369 newly diagnosed pediatric HHA patients in Korea, 71.3% had RBC membranopathies, 16.0% had hemoglobinopathies, 6.2% had RBC enzymopathies, and 6.5% had unknown etiologies. These data show the importance and necessity for the development and continuous improvement of the standardized diagnostic guidelines of HHA for clinical practitioners to suspect and diagnose HHA in Korean patients with anemia.
Patients with HHA may have similar clinical and laboratory findings. However, different therapeutic approaches may be required according to disease cause. Although splenectomy may be effective in the management of patients with HS, patients with hereditary stomatocytosis (HSt) do not benefit from splenectomy and are at increased risk of thromboembolic disorders after splenectomy [7, 8]. Thus, the RBC Disorder WP of the KSH had developed the Korean Standard Operating Procedures (SOPs) for the diagnosis of HHA in 2007 and had continuously updated the SOPs to ensure accurate and nationally standardized diagnostic methods and laboratory reports [9]. The initial Korean SOPs for the diagnosis of HHA were a step-by-step diagnostic process, including assessment of family history of anemia, gall stone or splenectomy, clinical examinations, and laboratory tests for anemia. These laboratory tests included complete blood count (CBC) with RBC index, peripheral blood morphology, iron study, bilirubin, lactate dehydrogenase (LDH), haptoglobin, antiglobulin test, osmotic fragility test (OFT), hemoglobin electrophoresis, globin gene analysis, RBC membrane protein analysis, and RBC enzyme analysis [9].
Subsequently, SOPs were updated by introducing more sensitive screening methods for RBC membranopathies [e.g., flow cytometric OF test (FOFT) and eosin-5-maleimide (EMA) binding test] and current methods for membrane protein or enzyme analysis [e.g., liquid chromatography-tandem mass spectrometry (LC-MS/MS), ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS), and high-performance liquid chromatography (HPLC)] [5, 9]. In addition, following recent advances in molecular diagnostic technologies, such as next-generation sequencing (NGS), the RBC Disorder WP of the KSH has developed an NGS gene panel for Korean HHA patients [10]. Considering the wider availability of gene testing in more laboratories in Korea, the panel included a proposal in the new Korean Clinical Practice Guidelines for the Diagnosis of HHA that NGS be performed prior to membrane protein or enzyme analysis by LC-MS/MS, UPLC-MS/MS, or HPLC techniques.
In this study, the authors attempted to update the SOP for HHA diagnosis on behalf of the RBC Disorder WP in KSH. Approaches for patients with anemia, suspected of HHA are described, including laboratory tests focused on HHA. We hope that this guideline may be helpful for clinicians in making diagnostic decisions for HHA patients in Korea.
GENERAL APPROACH TO THE PATIENT WITH HHA
History and clinical manifestations
The most common chief complaint of HHA is neonatal hyperbilirubinemia (within 48 hr of birth) requiring immediate medical intervention. With the exception of β-globin-associated HHA, most cases require more than just phototherapy, with some severe cases require exchange transfusion to avoid kernicterus [8]. The incidence of neonatal hyperbilirubinemia in glucose-6-phosphate dehydrogenase (G-6PD) deficiency is higher in male patients due to X-chromosome-related inheritance, but G-6PD deficiency should also not be ruled out in females because they can also present with mild symptoms through X-chromosome lyonization [11]. Intrauterine symptoms in fetuses with HHA are common. Intrauterine anemia, hyperbilirubinemia, intrauterine growth retardation, hydrops fetalis, and preterm labor have been reported as complications of HHA, especially in HS and enzymopathies [8, 12].
β-globin-related HHAs such as β-thalassemia or sickle cell disease usually present with anemia at 4 to 6 months of age [13]. Patients typically present with chronic HA, ranging from asymptomatic to compensated hemolysis to severe anemia, some requiring chronic transfusion therapy. Although the disease severity differs, most HHA patients need cholecystectomy or splenectomy [8]. Rarely, some neonates with HA develop jaundice that resolve in the first few months of life. These patients might have unstable hemoglobinopathy due to mutations in the γ-globin chain that resolves as fetal hemoglobin is replaced by adults [12, 14].
Some patients also develop hemolytic events, such as aplastic crisis that is commonly caused by parvovirus B19 infection [15]. Membranopathies, sickle cell disease, and enzymopathies such as G-6PD deficiency may be diagnosed incidentally by presenting with aplastic crisis events. Hemo-globinopathies, such as sickle cell disease and thalassemia, usually present as extensive extravascular hemolysis with increased blood viscosity due to excessive globin chain [16]. This typically leads to deep vein thrombosis, edema, pain, peripheral ulcers, and circulatory problems. Depending on the severity of hemolysis, patients may also manifest typical bony abnormalities and severe hepatosplenomegaly, which occur relatively earlier than do other HHAs [17].
The peripartum period can also yield pertinent diagnostic information. Specifically, the method of delivery (with or without instrumentation), history of maternal hemorrhage (e.g., vaginal or placental), evidence of fetal distress, and the presence of multiple gestations may all be crucial evidence. Further information regarding the pathology of the placenta and umbilical cord and its insertion may also be useful. Family history should also be determined when investigating causality of anemia in the newborn because certain conditions of autosomal recessive (e.g., Fanconi’s anemia), autosomal dominant or autosomal recessive [e.g., HS and hereditary elliptocytosis (HE)], and X-linked inheritance (e.g., G-6PD deficiency) may be disease etiologies. Less specifically, a history of relatives with anemia, jaundice, or splenectomy can also aid in directing further diagnostic evaluations.
Finally, the patient’s history is essential for diagnosis. Particularly, the gestational age and days of life at the time of presentation, ethnicity/race, and sex of the infant are all useful not only in determining a diagnosis of anemia, but also in establishing etiology such as in G-6PD deficiency and thalassemia. The diagnosis and etiology of neonatal and infancy anemia can often be determined with a thorough history taking; however, in certain situations, further diagnostic work-up may be required.
Physical examination
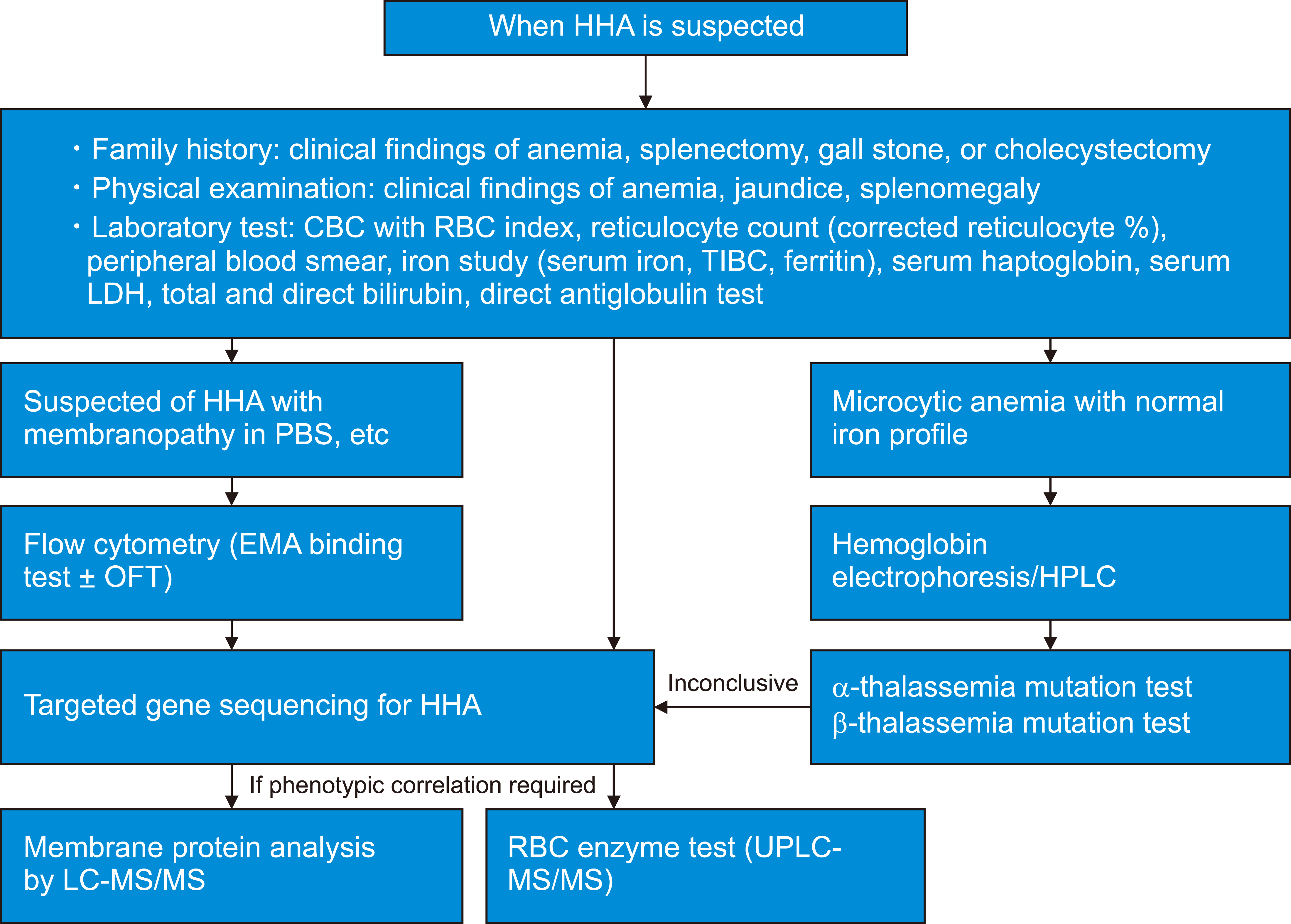
Intra- or extra-vascular premature destruction of RBCs results in anemia and an increase in production of unconjugated bilirubin, which leads to jaundice [18]. In the case of intravascular hemolysis, hemoglobin may be excreted into the urine (hemoglobinuria), leading to dark red-brown colored urine and therefore, HHA is suspected when the patient shows clinical findings of anemia, jaundice, and splenomegaly (Fig. 1). HHA should also be considered when neonates have jaundice requiring phototherapy or exchange transfusion. However, the severity of jaundice or hyperbilirubinemia does not predict the severity of the disease in the future [19].
Among HHA patients, those with membranopathies (HS or HE) generally show extravascular hemolysis. Although most patients with HS have mild HA, patients with autosomal recessive HS have severe anemia, which is transfusion dependent [19]. Further, although majority of patients with HS have mild to moderate splenomegaly [20, 21] and splenomegaly itself assists in the diagnosis of HS, it has little clinical significance [20, 21]. HS patients with well-compensated anemia are diagnosed later in life from the related cholelithiasis [14]. Hemoglobinopathies (thalassemia or sickle cell disease) generally show extravascular hemolysis. Patients with thalassemia major show typical clinical features of facial changes (enlarged maxillary sinuses) from hematopoiesis or blood cell production and such changes in the face promote infections of the ears, nose, and throat in patients with thalassemia major. Chronic transfusion-dependent anemia and iron overload, cardiomyopathy, diabetes mellitus, hypopituitarism, hypoparathyroidism, hypothyroidism, or testicular/ovarian failure are frequent in adults with thalassemia major [13].
Enzymopathies [G-6PD deficiency or pyruvate kinase (PK) deficiency] generally present with intravascular hemolysis [22]. In the case of G-6PD deficiency, intra- and extravascular hemolysis can occur. In patients with G-6PD deficiency, jaundice usually develops during the first postnatal day [19]. After infancy, most patients with G-6PD deficiency do not have anemia or jaundice at baseline, but hemolysis is triggered by oxidative stressors including medication, infections or fava beans. After exposure to oxidative stressors, acute HA develops with pallor, jaundice, fatigue, back pain, and red-brown urine in patients with G-6PD deficiency [19].
Meanwhile, patients with PK deficiency have differing clinical course. Some patients with PK deficiency have lifelong fully compensated or mild anemia, while others have transfusion-dependent anemia [1]. Hyperbilirubinemia in neonates with PK deficiency can be severe and require an exchange transfusion. Cholelithiasis or transfusion-dependent iron overload are common in cases of severe phenotypes of PK deficiency and the degree of hemolysis can be exacerbated during infections or pregnancy [23]. Splenomegaly is not prominent, and the benefits of splenectomy vary by case [12]. Further specific findings of physical examination of patients with HHA are shown in Table 1 [18, 24].
Laboratory tests for screening
Laboratory tests to be performed in patients suspected of HHA are shown in Fig. 1. Testing commonly starts from complete blood count (CBC) and reticulocyte count. The WHO criteria for anemia vary with age and sex, as shown in Table 2 [25]. The corrected reticulocyte count (CRC, %) and reticulocyte production index (RPI) are calculated as follows:
In general, RPI reflects the erythropoietic activity of bone marrow, with RPI >3 indicating an appropriate bone marrow response and RPI <2 indicating an inadequate response to correct anemia [27]. The severity of anemia reflects the clinical severity of hemolytic diseases [28], and individuals may show normal hemoglobin levels with reticulocytosis, suggesting compensated hemolysis or reticulocytopenia despite hemolysis in other cases [28]. Red cell indices can be used for differential diagnosis. HS patients have elevated mean corpuscular hemoglobin concentration (MCHC) from relative dehydration, suggesting the presence of spherocytes with the loss of membrane surface area relative to intracellular volume. In HA patients with low mean corpuscular volume (MCV) and mean corpuscular hemoglobin, the work-up of thalassemia should follow once iron deficiency is ruled out. Markers of hemolysis, including elevated lactate dehydrogenase (LDH), urinary and fecal urobilinogen, unconjugated hyperbilirubinemia, and decreased serum haptoglobin are present in varying degrees in HA [29]. Direct antiglobulin testing (direct Coombs’ test) is performed to detect antibodies or complement system factors that are bound to RBC surface antigens. Patients who test positive are suspected for HA caused by immune-mediated causes rather than intrinsic defects of RBC membranes or enzymes [22].
Peripheral blood smear
A peripheral blood smear (PBS) can be used for the differential diagnosis of nonimmune HA. Spherocytic red cells are predominant red cell-shaped abnormalities in HS [30]. Defects in the RBC membrane due to various genetic alterations lead to spherocyte formation [30]. Although spherocytes are predominant, additional red cell morphologies can also be observed [14]. Anisocytosis is noted in patients with ANK1-associated HS, mushroom-shaped red cells are characteristic of band 3 (SLC4A1) deficiency, and acanthocytes/echinocytes are associated with SPTA1-associated HS [30]. Ovalocytes and ovalostomatocytes have been observed in EPB42-associated HS [30]. Other inherited red cell membrane disorders include HE and HSt. Mild HE is characterized by remarkable elliptocytosis without HA, whereas moderate to severe HE commonly presents with fragmented red cells associated with HA [14].
Meanwhile, HSt presents with a high number of stomatocytes, but the red cell morphology is relatively normal [14]. In addition, red cell inclusions can aid in the diagnosis of non-immune HA. Heinz bodies are commonly observed in abnormalities of alpha and beta hemoglobinopathies [31]. Hypochromic anemia is also a typical finding of unstable hemoglobin in PBS. Meanwhile, although PBS findings are not critical for diagnosis, they may be helpful in disorders of erythrocyte metabolism. For example, basophilic stippling is characteristic of pyrimidine 5’ nucleotidase (P5N) deficiency [31]. Occasionally, neonatal HA causes G-6PD deficiency or PK deficiency to overlap with infantile pyknocytosis, which can confuse the differential diagnosis [21, 32]. Postponing morphology tests can sometimes be considered until at least 6 months of age [21].
Osmotic fragility test and eosin-5-maleimide binding test by flow cytometry
OFT has been performed for HS screening to testing the capability of RBCs to incorporate hypotonic solutions. However, because of its low sensitivity, specificity, and lack of standardization, OFT is not recommended, and FOFT has been introduced [33-35]. FOFT measures the difference in RBC at baseline with normal saline and after incubation with hypotonic saline. Although protocols vary among laboratories, FOFT have been shown to have higher sensitivity and specificity than OFT [34, 36]. Eosin-5-maleimide (EMA) is thought to bind to membrane proteins (band 3, CD47, Rh protein, and Rh glycoprotein) on RBCs, which are indirectly associated with the spectrin-based RBC cytoskeleton [35, 37]. A reduction in fluorescence intensity of EMA-labeled RBCs on flow cytometry indicates a membrane protein-associated RBC disorder. Patients with a family history of HS, typical clinical features (splenomegaly), and laboratory features (spherocytes, increased MCHC, increase in reticulocytes) do not require additional tests according to the guidelines for the diagnosis of HS [21].
However, when the diagnosis is equivocal, cryohemolysis and EMA binding tests are recommended for screening. Although the EMA binding test is highly sensitive for HS, Southeast Asian ovalocytosis (SAO), cyrohydrocytosis, congenital dyserythropoietic anemia (CDA) type II, and hereditary pyropoikilocytosis can also present with fluorescence intensity in the same range as HS [35, 38]. Thus, the clinical presentation and PBS results should be reviewed in conjunction. Some HS patients with isolated ankyrin deficiency exhibit normal fluorescence. The cutoff value for HS varies by laboratory because of the differing EMA preparations, and thus, it should be established according to the testing site [39]. Further, there exists a gray zone between normal controls and HS [40], and family studies and other tests may be required. Both FOFT and EMA testing are available in several laboratories in Korea [33, 39], and they can be ordered for patients suspected of HS.
High-performance liquid chromatography and hemoglobin electrophoresis
Hemoglobinopathies are screened by either HPLC or isoelectric focusing in the United States [41]. In Korea, hemoglobinopathies are evaluated using HPLC or hemoglobin electrophoresis, which is available in many laboratories [42]. Hemoglobin fractions are separated based on their ionic interaction, and each hemoglobin has its own retention time, which is measured from the time of sample injection into the HPLC to the maximum point of each peak. Unknown hemoglobin fractions are identified by comparison with known hemoglobin retention times [43]. With electrophoresis, charged molecules are separated by their electrophoretic mobility, either in alkaline or acidic pH conditions. In alkaline electrophoresis (pH 8.6–9.2) with cellulose acetate, more negatively charged hemoglobins move to the anode faster, and the separation of HbA, HbA2, HbF, HbS, and HbC is reliable. In acid electrophoresis with citrate agar, HbS can be separated from HbD and HbG, and HbC from HbE and HbO [44, 45]. Capillary zone electrophoresis (CZE) is performed in an alkaline buffer (pH 9.4) where hemoglobin fractions migrating from the anodic end of the capillary appear in specific zones to the cathodic end, and detection occurs at 415 nm [46]. Although the reference range may vary by method, it is usually 96% for HbA, <1% for HbF, and 2.5–3.5% for HbA2. In β-thalassemia, HbA is decreased and HbA2 is slightly increased (<10%), with variably increased HbF. An HbA2 level above 10% suggests variant hemoglobin rather than β-thalassemia. The decrease in HbA levels depends on the genetic variations in β-thalassemia. Meanwhile, in α-thalassemia, HbA2 is normal or slightly decreased, and HbH or HbBart can be present in newborns. In Korea, abnormal CZE or HPLC results are confirmed by genetic testing, usually with Sanger sequencing and/or multiplex ligation probe amplification (MLPA) [42].
DIAGNOSTIC LABORATORY TESTS FOR HHA
RBC membranopathies
RBC membrane disorders can be categorized into two groups: 1) RBC disorders caused by membrane structural defects (e.g., HS, HE, hereditary pyropoikilocytosis, and SAO) and 2) RBC disorders caused by altered membrane transport function (e.g., HSt, familial pseudohyperkalemia, and cryohydrocytosis) [47, 48]. Although genetic testing is an important tool for differential diagnosis [49, 50], approximately 50% of cases are undiagnosed despite NGS testing [47]. Further, several atypical cases of RBC membrane structure defects may need quantification of the membrane proteins for mutations not consistent with the phenotype [51] and for diagnostic confirmation according to the ICSH guidelines [52]. Quantification of RBC membrane proteins has been traditionally performed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) [47, 52, 53]. However, more advanced mass-spectrometry-based techniques with matrix-assisted laser desorption/ionization-time of flight mass spectrometry [54] or LC-MS/MS [54, 55] have been used recently, but these are currently unavailable in Korea. Genetic testing can be considered in cases of suspected RBC membranopathy.
HS not only has highly variable phenotypes, but is also associated with genetic variants of cytoskeleton and transmembrane protein quantitative deficiencies, including ankyrin, erythrocytic α- and β-spectrin chains, band 3 protein, and protein 4.2 [29, 47]. The most frequently reported variants in HS are deficiency of ankyrin and band 3 proteins [52, 56]. Approximately 8–11% of patients have no detectable reductions in membrane proteins [52]. Membrane protein quantification by SDS-PAGE can be helpful in differentiating CDA type II from HS [47].
HE has defects in the proteins of the junctional complex (protein 4.1) and spectrin chains, which are caused by autosomal dominant variants in the EPB41, SPTA1, and SPTB genes [47]. Protein 4.1 deficiency due to EPB41 mutation can be detected in HE by simple membrane protein quantification [53]. However, some patients with SPTA1 or SPTB mutations do not have quantitative deficiencies. In these patients, the pathogenic mechanism of spectrin defect is due to tetramerization impairments, in which two-dimensional gel electrophoresis of tryptic spectrin digest is needed [52].
RBC enzymopathies
Classical spectrophotometric methods to measure RBC enzyme activity involve specific reactions set up for each RBC enzyme, and changes in cofactor substrates (NAD+, NADP+, NADH, or NADPH) are measured at 340 nm [57]. These classical methods are generally labor intensive and time consuming because they require multiple reactions for each specific enzyme, and each enzyme must be measured separately. To overcome these disadvantages, LC-MS/MS-based multiplex RBC enzyme assays have been developed to measure the activities of enzymes implicated in RBC enzymopathies.
The current mainstream method for measuring RBC enzyme activities in Korea is an ion-pairing, UPLC-MS/MS method [58], which measures the activities of six major RBC enzymes, namely, G-6PD, PK, P5N, hexokinase (HK), triosephosphate isomerase (TPI), and adenosine deaminase (AD). The enzyme activities of five of these six enzymes (G-6PD, PK, P5N, HK, and AD) are measured in a one-step process. The respective enzyme reaction products, namely, 6-phosphogluconate, pyruvate, cytidine, C5- glucose-6- phosphate, and inosine, are directly measured by LC-MS/MS. For TPI, the primary reaction product (dihydroxyacetone phosphate) is indistinguishable from the substrate (glyceraldehyde- 3-phosphate) on mass spectrometry. Therefore, a secondary reaction with α-glycerophosphate dehydrogenase is performed, which allows the measurement of the secondary reaction product (i.e., glycerol-3-phosphate) via LC-MS/MS. RBC enzyme activity results are reported in mg/min/gHb for G-6PD and PK and in µg/min/gHb for P5N, HK, TPI, and AD. Current reference levels, evaluated from 83 normal controls, are 0.96–3.14 mg/min/gHb for G-6PD; 1.01–3.01 mg/min/gHb, PK; 9.5–35.7 µg/min/gHb, P5N; 132–532 µg/min/gHb, HK; 423–961 µg/min/gHb, TPI; and 86.5–660.9 µg/min/gHb, AD, respectively.
The limitation of the current UPLC-MS/MS RBC enzyme activity assay is that it covers only six enzymes. Although deficiencies in the aforementioned six enzymes contribute to the majority of RBC enzymopathy cases, other enzymes including adenylate kinase, aldolase, phosphofructokinase, phosphoglycerate kinase, glucose-6-phosphate isomerase, glutathione reductase, and glutathione synthetase are also known to be associated with RBC enzymopathies [59]. Thus, clinicians should be clinically aware of which RBC enzymes are being tested and should not completely rule out any untested diagnoses. Furthermore, there are some notes of caution when interpreting enzyme assay results [31]. The in vivo environment may differ to laboratory-prepared, in vitro enzyme reactions. False masking of enzyme activity deficiency can occur under contamination by transfused normal RBCs or by glycolytic enzymes in leukocytes, highlighting the importance of leukocyte depletion in the sample preparation step. Moreover, the reported results are the averages of RBC enzyme activities, which may not accurately demonstrate activities in RBC subpopulations. Thus, possible variables such as reticulocytosis should be identified and considered before interpretation of RBC enzyme activity results.
Hemoglobinopathies
A retrospective epidemiologic study of HHA in Korea showed that majority of hemoglobinopathies were β-thalassemia minor (83.1%) and α-thalassemia minor (15.3%) [5]. In the case of β-thalassemia, mutation is mainly a single nucleotide variation in the HBB gene, and is detectable by the sequence analysis. However, it can also include variants in noncoding regions and, rarely, large deletions. If only one or no pathogenic variant is identified through HBB sequencing, deletion/duplication analysis is performed. In contrast, in the case of α-thalassemia, approximately 85% of all causative mutations are deletions in the HBA1 or HBA2 gene. Therefore, MLPA should be performed first, and sequence analysis of HBA1 and HBA2 can be performed if a common deletion is not identified. However, because MLPA is not covered by health insurance, MLPA is not routinely performed, and thus, the incidence rate of HHA may be underestimated in Korea [5].
Genetic testing for HHA
Accurate diagnosis of HHA is challenging because the clinical features may overlap even when the etiology is completely different. Thus, genetic testing is recommended if (i) routine studies have failed or achieved inconclusive to identify the causative mechanism; (ii) transfusions have recently been administered, leading to confounding biochemical and other testing due to mixed RBC populations; and (iii) the patient is a newborn [48]. As various genetic mechanisms regulating the function of RBCs have been elucidated [48, 60], genetic testing is increasingly being used for the confirmatory diagnosis of HHA in Korea. Until recently, molecular diagnosis has been primarily performed using Sanger sequencing techniques, typically by sequencing single genes [61]. Targeted gene panels have been developed and applied to routine molecular diagnosis for undiagnosed patients and family members of HHA [61, 62]. In addition, they are preferred over whole exome or whole genome sequencing due to lower cost and turn-around time, decreased complexity of data analysis, better coverage over the regions of interest, and reduced incidental findings [63].
The RBC Disorder WP of the KSH has developed a targeted gene panel that covers genes associated with RBC membranopathy, enzymopathy, and other diseases that have overlapping features with HHA (Table 3). NGS testing can be considered prior to RBC enzyme or membrane protein analysis because of the wider availability of genetic testing in Korea. Phenotypic analysis can then be used to support the molecular findings. However, the NGS panel of RBC Disorder WP or many other laboratories do not include testing for hemoglobinopathies because specific considerations are necessary regarding detection of large deletion/duplication, variants in introns, and copy number variations. All genetic tests should be designed to cover the major coding regions and intronic flanking regions of the gene. Each laboratory should validate its performance before clinical application [64]. Given that genetic tests have limited capability to identify variants of uncertain significance or to detect variants that do not explain the underlying phenotype, the results should be reviewed by professional geneticists.
CONCLUSION
HHAs are challenging to diagnose because of the many confounding factors in the clinical setting, thus confusing clinicians. However, technological advances have enabled the elucidation of the pathophysiologies of many HHAs, and diagnostic tests are being updated. In addition, new technologies such as LC-MS/MS or NGS cover many diseases through a single test, enabling clinicians to gather more information simultaneously. However, accurate history taking and physical examination remain the most important diagnostic strategy for HHA. For patients suspected of having HHA, family history, past medical history, clinical presentation, and physical examination should be considered in the assessment in addition to sex and race, and the next step can be decided according to the PBS results. If spherocytes or other features of membranopathy are present, the EMA-binding test should be performed first. If PBS shows features suspected of thalassemia, MLPA or genetic testing should be selected for hemoglobin genes. If PBS findings are inconclusive, an enzyme test or NGS should be considered. However, there are limitations in current SOPs, and many HHAs cannot be diagnosed through the methods mentioned earlier. The recommended tests are now commercially available in Korea, and the RBC Disorder WP and the KSH will make any effort to update and improve the diagnostic SOPs of HHA. We hope that this report will be helpful in the diagnostic workup for patients with HHA in Korea.
 XML Download
XML Download