

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Gout is an autoinflammatory disorder induced by deposition of monosodium urate (MSU) crystals in the articular joints and periarticular structures and is characterized by abrupt, intermittent, and self-limiting episodes of painful arthritis [1,2]. In addition, nodules called tophi can develop at diverse locations such as ear helix, malleolus of the ankle, and olecranon of the elbow in advanced gout. Some patients might experience impaired renal function or chronic renal disease induced by deposition of MSU crystals. Gout was historically documented by the Egyptians in 2640 BC and was described as ‘arthritis of the rich’ (Greek: podogra) by Hippocrates (460-377 BC) [3]. Hyperuricemia or increased concentration of serum uric acid is the most crucial trigger factor and a prerequisite for development of gout. Although acute arthritis by intraarticular injection with sodium urate microcrystals was observed through experiments in 1899 [4], and MSU crystals were identified in the synovial fluid of patients with gouty arthritis by McCarty and Hollander in 1961 [5], the precise mechanism by which MSU crystals cause the acute inflammatory response of gout has only recently been unraveled.
Recent advances suggest that activation of an innate immune system response to MSU crystals triggers the inflammation in gout. Abundant proinflammatory cytokines and chemokines such as interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), and IL-8 were identified in the inflammatory joints of patients with gout [6]. Recent studies demonstrated that IL-1β is a key mediator of the gout-induced inflammatory response to MSU crystals [7]. Activation of the NACHT (neuronal apoptosis inhibitory protein [NAIP], MHC class II transcription activator [CIITA], incompatibility locus protein from Podospora anserina [HET-E], and telomerase-associated protein [TP1]), leucine-rich repeat (LRR), and pyrin domain (PYD)-domains-containing protein 3 (NLRP3) inflammasome is an essential step for conversion of the inactive form of IL-1β, pro-IL-1β, to mature IL-1β by inflammatory cells including macrophages, monocytes, and dendritic cells following exposure to various pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [8]. Mononuclear phagocytes recognize that MSU crystals playing a role as a DAMP initiate the production and release of inflammatory cytokines, especially the IL-1β family of cytokines such as IL-1β and IL-18, through activation of the NLRP3 inflammasome [7]. For treatment of gout, IL-1 inhibitors such as IL-1R antagonist (anakinra) and monoclonal anti-IL-1β antibody (canakinumab) targeting for IL-1β show therapeutic efficacy in patients with acute and chronic gouty arthritis [9]. This suggests the NLRP3 inflammasome pathway regulating IL-1β and IL-18 among the IL-1 superfamily and IL-1-linked inflammatory cascades as a key therapeutic target for management of gout. In this review, we discuss the mechanism of NLRP3 inflammasome activation by endogenous or exogenous danger signals (i.e., PAMPs and DAMPs) and the role of the NLRP3 inflammasome in response to MSU crystals in the pathogenesis of gout.
MAIN SUBJECTS
Overview of the NLRP3 inflammasome
Inflammation is usually caused by diverse insults such as extracellular infectious pathogens, intracellular stress molecules derived from damaged tissues or organs, or environmental irritants. In the human body, appropriate mechanisms for recognizing and processing these insults and the effectors to repair and control tissue injury are initiated. These cellular and molecular pathogens and damaged molecules are first recognized by innate immune systems through a repertoire of germline-encoded receptors called pattern-recognition receptors (PRRs) such as the nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), the retinoic acid-inducible gene I-like receptors (RLRs), and the Toll-like receptors (TLRs) [10]. PPRs typically recognize two different danger signals, PAMPs with conserved microbial signatures and DAMPs derived from tissue damage and cellular apoptosis (Table 1).
Among PRRs, the NLRs are cytosolic receptors with tripartite architecture that share a LRR at C-terminal, a central nucleotide domain referred to as NACHT, and N-terminal effector domains including PYD, caspase activation and recruitment domain (CARD), or baculovirus inhibitor of apoptosis repeat (BIR) domain and ultimately function as molecular platforms such as inflammasomes and NOD signalosomes that sense cytoplasmic signaling by the presence of PAMPs and DAMPs [11]. NLRs are divided into three subfamilies: the 14 members of the PYD-containing NLRP3 (NLRP1 to NLRP14), the five members of the NODs (NOD1 to NOD5) and CIITA, and the remaining NLR members (IPAF and the BIR-containing NAIP). NLRs generally contain three distinct domains such as LRR, NACHT, and PYD. The LRR domain has diverse functions including pathogen sensing and autoregulation of NLRs, although the mechanism for interaction between the LRR and ligands has not been clearly defined. The NACHT domain plays a central role in oligomerization of NLRs. The PYD or CARD, as N-terminal effector domains, is responsible for signal transduction to downstream targets, triggering a process of inflammatory caspase activation through inflammasome complex.
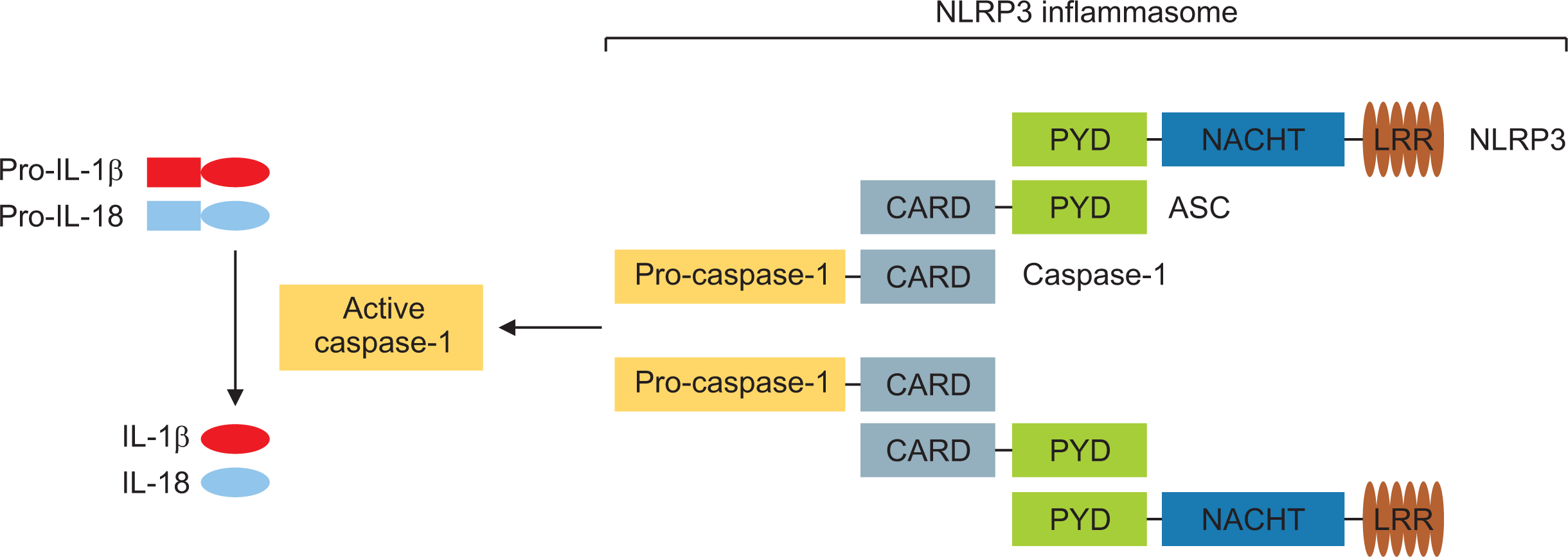
The inflammasome is an intracellular multi-protein signaling platform that has cytosolic PRRs, including NLRs to endogenous and exogenous pathogens, and plays a crucial role in activation of the innate immune system through activation of inflammatory caspases, maturation of pro-IL-β, and secretion of IL-1β [12]. Among several members of NLRs, NLRP3, which was initially described as NALP3 and cryopyrin, has been mostly well defined (Figure 1). The NLRP3 inflammasome consists of sensor NLRP3, the adaptor protein ASC, and inflammatory pro-caspase-1. Following stimulation with multiple NLRP3 activators, including PAMPs and DAMPs, directly through the LRR domain or other mediators, oligomerized NLPR3 protein interacts with the PYD domain of the ASC protein. Subsequently, ASC recruits pro-caspase-1 through a CARD. Activated caspase-1 cleaves precursors of the proinflammatory cytokines pro-IL1-β and pro-IL-18 to mature IL-1β and IL-18, ultimately leading to secretion of these inflammatory cytokines.
Mechanism for the signaling pathway regulating NLRP3 inflammasome activation
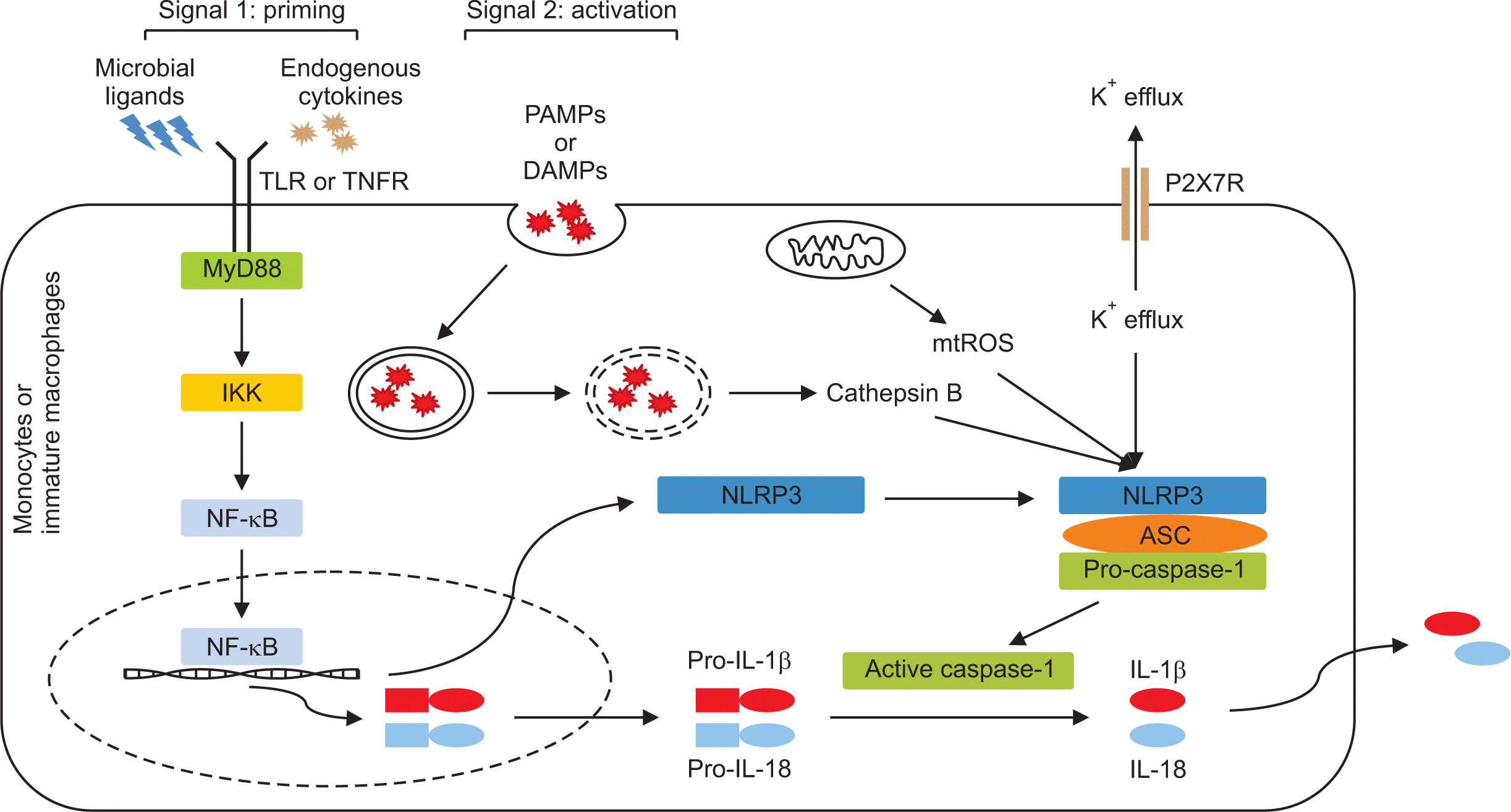
1) Signal 1: NLRP3 priming
NLRP3 inflammasome activation in macrophages is either not at all or weakly detected under stimulation of NLRP3 activators alone, whereas pretreatment with microbial ligands or endogenous cytokines strongly induces NLRP3 inflammasome activation [13-15]. This pretreatment is called the “priming signal” and provides the first signal to trigger the initial step for NLRP3 inflammasome activation in innate immune cells (Figure 2). Cell priming induced by multiple TLR ligands such as lipopolysaccharide (LPS) and lipopeptide, and by diverse endogenous proinflammatory cytokines through cytokine receptors for TNF or IL-1β could enhance the expression of NLRP3 inflammasome components such as NLRP3 and the inactive form of pro-inflammatory cytokines such as pro-IL-1β and pro-IL-18, which are the main caspase-1 substrates, through nuclear factor-κB (NF-κB) pathway activation at the transcriptional level [13,16].
Evidence supports the idea that several pathways are involved in the NLRP3 priming process through transcriptional and post-translational modifications over times [17]. The first pathway takes several hours (>3 hours) to license NLRP3 after binding of TLR and its agonists and is referred to as ‘transcriptional’ or ‘late’ priming [13]. It engages the myeloid differentiation primary response protein 88 (MyD88)/IL-1 receptor-associated kinase 4 (IRAK4), the TIR-domain-containing adaptor protein inducing IFN-β (TRIF), and the Fas-associated protein with death domain (FADD), which activates transcriptional factor NF-κB [18]. The expression of NLRP3 protein is insufficient for NLRP3 inflammasome activation, and pro-IL-1β is not presented in resting macrophages. Therefore, it is necessary to transcriptionally upregulate the expression of free cytosolic NLRP3 protein maintained in an inactive ubiquitinated state and pro-IL-1β through the priming step. Recent studies demonstrated that macrophages with overexpression of NLRP3 using a retroviral vector markedly induced NLRP3 inflammasome activation without priming [13,19], suggesting that priming for NF-κB-dependent transcription upregulation of NLRP3 is a crucial step for induction of NLRP3 inflammasome activation. Unlike transcriptional priming, TLR stimulation quickly induces priming of NLPR3 within minutes (10~30 minutes) without processing of protein synthesis such as NLPR3, which is referred to as ‘non-transcriptional’ priming [20,21]. This implies that the basal level of NLRP3 required for inflammasome activation is present in sufficient quantity, and this early priming mechanism might be dependent on post-translational modification. The adaptor MyD88 and IRAK1/4 pathway is engaged following stimulation with TLR2 and TLR4 agonists in this non-transcriptional priming, resulting in conversion of unprimed NLRP3 to primed NLRP3.
2) Signal 2: NLRP3 inflammasome activation
After priming the licensing of NLRP3, diverse danger signals such as DAMPs and PAMPs induce assembly of NLRP3 and NLRP3 inflammasome activation (Figure 2). This process is referred to as signal 2. Activation of primed NLRP3 responses to diverse stimuli such as cellular stresses and particulate crystals. First, some NLRP3 agonists such as pore-forming toxins, complement membrane attack complexes, and extracellular ATP via membrane P2X7 receptors, which contribute to plasma membrane permeability to ions, trigger NLRP3 activation [22-24]. In addition, phagocytosis by crystal particulates including asbestos, silica, cholesterol crystals, MSU, and ASC specks induces phagolysosome rupture, leading to reactive oxygen species (ROS) generation and Cathepsin B release, ultimately activating NLRP3 [25,26]. It is unlikely that primed NLRP3 directly senses a variety of NLRP3 activators located in the extracellular space in the process of NLRP3 inflammasome activation. It has been proposed that NLRP3 inflammasome activation triggered by a variety of NLRP3 stimulators is dependent on secondary signaling mechanisms such as K+ efflux and Ca2+ mobilization, lysosomal destabilization, and ROS generation (Table 2).
(1) Ion channel model: K+ efflux and Ca2+ mobilization
K+ efflux or low intracellular K+ concentration caused by numerous NLRP3 activators has been recognized as an upstream phenomenon in NLRP3 inflammasome activation. In 1994, before the concept of the NLRP3 inflammasome was introduced, ATP and nigericin were shown to induce a net decrease in intracellular concentration of K+ that plays a crucial role in maturation and release of IL-1β from LPS-stimulated macrophages [27]. Accelerated processing of IL-1β and caspase-1 maturation from Bac1 murine macrophages stimulated with ATP and nigericin was found to be dependent on intracellular K+ loss [28]. Furthermore, IL-1β maturation in human monocytes and murine macrophages stimulated by nigericin, ATP, and some bacterial infections such as that of E. coli. was blocked by addition of 130 mM extracellular potassium [29].
Pannexin 1, a transmembrane channel protein that delivers ions and extracellular signaling stimuli, is activated through binding of ATP to P2X7R, ultimately causing the release of IL-1β. ATP triggers pore formation of pannexin 1, which functions as a membrane channel and is required for processing of caspase-1 and release of mature IL-1β [30]. The P2X7R-gated ion channel is required for ATP-mediated IL-1 maturation and release through rapid K+ efflux from the cytoplasm [31]. NLRP3-activating bacterial endotoxins such as Leucocidin A/B, Streptolysin O, and α-Hemolysin appear to contribute to NLRP3 inflammasome activation through K+ efflux by pore formation [32]. Thus, pore formation in the transmembrane due to bacterial toxin could provide possible evidence for NLRP3 activation induced by K+ efflux. In addition, phagocytosis of particulate matters such as SiO2, Al(OH)3, and CPPD crystal enhances K+ efflux, resulting in NLRP3 activation [33]. This suggests that some particulate molecules that pass through the cell membrane can activate NLRP3 through the pore or channel. Although the channel model related with K+ efflux could not wholly account for NLRP3 activation, lowering intracellular K+ level has been proposed as a crucial pathway for NLRP3 inflammasome activation after exposure to NLRP3 stimuli including bacterial toxins, ATP released from cellular injury or necrosis, and phagocytosis of some particulates.
It has been recognized that Ca2+ signaling is also involved in NLRP3 inflammasome activation. Increased intracellular Ca2+ mobilization leads to the NLRP3 inflammasome mediated through the murine calcium-sensing receptor [34]. Consistently, intracellular Ca2+ level is increased by release from both extracellular and intracellular pools in LPS-primed bone marrow-derived macrophages (BMDMs) stimulated with extracellular ATP through P2X7R [35]. In addition, thapsigargin, a potent inhibitor of the sarco/endoplasmic reticulum Ca2+-ATPase, inhibits cytosolic Ca2+ mobilization through blockage of extracellular Ca2+ entry, which then attenuates assembly and activation of the NLRP3 inflammasome. The precise mechanism of intracellular Ca2+ mobilization in NLRP3 inflammasome activation has not been clearly defined. Ca2+ mobilization is tightly regulated at low cytoplasmic Ca2+ levels in an unstimulated state. If increased cytosolic Ca2+ mobilization from the extracellular space and endoplasmic reticulum triggers various cellular responses including cell proliferation, cellular metabolism, and apoptosis, cytosolic Ca2+ released from the cytoplasm or the endoplasmic reticulum enters the mitochondria to maintain intracellular Ca2+ homeostasis [36]. In other words, it is presumed that excessive cytosolic Ca2+ storage in the mitochondria results in mitochondrial damage and ultimately mitochondrial-induced ROS generation, which is a crucial mediator responsible for NLRP3 activation [37].
(2) Lysosomal destabilization and the cathepsin B model
An alternative mechanism is that lysosomal destabilization and rupture following phagocytosis of NLRP3 stimuli such as crystals or live pathogens trigger NLRP3 inflammasome activation. Lysosomal destabilization was first shown to contribute to NLRP3 inflammasome activation through Amyloid-β [38]. Cathepsin B released from lysosomal rupture of phagolysosomes into the cytoplasm seems to be a key molecule in regulation of NLRP3 inflammasome activation. Impaired clearance of particulate matters such as silica crystals and aluminum salts triggers lysosome rupture and release of cathepsin B, triggering NLRP3 inflammasome activation [39]. Supporting the evidence of the role of cathepsin B in NLRP3 activation, cathepsin B inhibitor (CA-074-Me) markedly inhibited NLRP3 inflammasome activation and synthesis and release of IL-1β from murine macrophages stimulated with silica crystals or alum [39,40]. In addition, lysosomal leakage for phagocytosis of β-hemolysin of group B Streptococcus and adenovirus leads to NLRP3 inflammasome activation [41,42]. However, the blockade of NLRP3 activation by cathepsin B inhibitors might be due to the paracrine effect or regulation of other members of the cathepsin family related to NLRP3. Interestingly, Muñoz-Planillo et al. [33] found that NLRP3 activation induced by phagocytosis of particulate matters was dependent on disturbance of the lysosomal membrane, together with K+ efflux, which has been recognized as a specific upstream regulator of NLRP3 inflammasome. However, there is a need to elucidate the mechanism for particulate matter-related lysosomal membrane damage and K+ efflux for NLRP3 inflammasome activation.
(3) ROS model
ROS generated around inflammasomes have been considered as a potent mediator for NLRP3 inflammasome activation. Mitochondria are key players that generate and release ROS during the process of oxidative phosphorylation. There is evidence that ROS released from the mitochondria positively regulates NLRP3 inflammasome activation [43,44]. Numerous NLRP3 stimulators such as viral and bacterial microorganisms, ATP, hyaluronan, glucose, and diverse particulate molecules trigger generation and release of mitochondrial ROS in various types of cells [8,12]. Mitochondrial dysfunction and apoptosis induced by multiple NLRP3 agonists including ATP, staurosporine, or nigericin lead to mitochondrial ROS generation and release of oxidized mitochondrial DNA (mtDNA) into the cytosol, resulting in NLRP3 inflammasome activation and secretion of IL-1β [45]. Several studies found that mutual interaction between mitochondrial ROS and K+ efflux (or low intracellular K+ level) caused by stimulation with Aspergillus fumigatus and Candida albicans contributed to NLRP3 inflammasome activation and IL-1β release [46,47], although the relationship between the two models for NLRP3 inflammasome activation is not clearly understood.
The exact mechanism through which ROS senses and interacts with NLRP3 has not been elucidated. Recent evidence suggested that NIMA-related kinase 7 (NEK7) is an essential component for NLRP3 inflammasome activation. NEK7 is bound to the leucine-rich repeat domain of NLRP3 and is markedly activated downstream of mitochondrial ROS [48]. Nigericin treatment in RAW264.7 cells primed with LPS leads to a shift of NLRP3 to higher molecular mass fractions, but this shift was blocked in knockdown murine macrophages transfected with NEK7 siRNA, suggesting that direct binding of NLRP3 and NEK7 is dispensable for NLRP3 oligomerization and activation during NLRP3 inflammasome activation. It seems that NEK7 acts as a sensor of mitochondrial ROS rather than NLRP3 itself. However, another study found that NEK7 acts as an NLRP3-binding protein that is regulated by K+ efflux but not ROS to control NLRP3 oligomerization and activation [49]. Nek7−/− macrophages showed marked depletion of caspase-1 and IL-1β expression in response to NLRP3 stimuli such as ATP, nigericin, alum, and CPPD, and NLRP3-NEK7 interaction is significantly blocked by the presence of KCl that inhibits potassium efflux. Thus, mitochondrial ROS and/or K+ efflux can be considered common mediators that activate the binding of NEK7 and NLRP3, ultimately triggering NLRP3 inflammasome activation and IL-1β secretion. Further evidence provides the molecular mechanism for oxidative stress-related NLRP3 inflammasome activation.
Thioredoxin-interacting protein (TXNIP) is an inhibitory molecule to thioredoxin (TRX) that regulates the level of ROS, blocking tissue injury from oxidative stress [50,51]. Considering the role of TXNIP in NLRP3 inflammasome activation, TXNIP was identified as an NLRP3 binding protein and is constitutionally bound to TRX in the unstimulated state. ROS induce the dissociation of TXNIP from TRX and binding to NLRP3, leading to ROS-sensitive NLRP3 inflammasome activation [52]. LPS-primed Txnip−/− BMDMs stimulated with MSU, ATP, or R-837 impaired the maturation of caspase-1 and IL-1β, suggesting that TXNIP is essential for NLRP3 inflammasome activation. Therefore, TXNIP is one of the key molecules for NLRP3 inflammasome activation in an ROS-dependent manner via multiple NLRP3 activators and is considered a therapeutic approach for diseases related to NLRP3 inflammasome.
The role of NLPR3 inflammasome in the pathogenesis of gout
1) Uric acid as a danger signal in gouty inflammation
Uric acid (2,6,8-trihydroxypurine) is the endogenous end product of catabolic metabolism of dietary purine nucleotides during oxidation of purine bases catalyzed by xanthine oxidase [7]. In particular, uric acid is the end product of purine metabolism in humans because of a defective mutation in the uricase gene. Uric acid can present in the body in both soluble and crystalline form. Uric acid maintains a dissolved state at serum concentrations less than 7.0 mg/dL in males and 6.0 mg/dL in females, but it can be crystalized at higher concentrations of uric acid (hyperuricemia) exceeding physiological saturation [2]. Hyperuricemia is a prerequisite for development of gout, although it does not necessarily progress to gout.
Microbial components related to bacterial, viral, and fungal infection act as danger signals that activate the immune systems and promote inflammatory response in innate immune cells [32,53]. In the absence of infection, some ill-defined danger signals released from dying or damaged cells alarm the host to cell damage and lead to an inflammatory response that urges initiation of the host defense mechanism and tissue repair. Shi et al. [54] found that uric acid was identified as an endogenous danger signal released from dying cells to activate an immune response through its adjuvanticity effect. Crystallization is required to produce the inflammatory properties of uric acid. Diverse humoral, environmental, and cellular factors related with MSU crystallization include body temperature, pH, serum IgM antibody, and cartilage matrix components in synovial fluid, other than excessive concentration of uric acid [1,55]. In particular, MSU crystallization occurs when extracellular uric acid in the condition of reduced uric acid solubility comes into contact with a high concentration of free sodium [55]. MSU crystallization occurs through this process, and it is considered a biologically active substance with the adjuvant effect of uric acid. Thus, uric acid released from cell death or damaged cells acts as a danger signal of an inflammatory response through the formation of MSU crystals.
2) NLRP3 inflammasome-mediated inflammation in gout
It has been more than a century since uric acid was identified as the cause of gout. More recently, the mechanism of action by MSU crystal-induced inflammation in gout has been elucidated. The pathogenic hallmark of gouty inflammation is that mononuclear phagocytes in the response to exposure to MSU crystals trigger the production of a proinflammatory cytokine, IL-1β, playing a crucial role in the gouty attack [56,57]. Shi et al. [54] demonstrated that uric acid was a potent endogenous danger-signaling molecule released from dying or damaged cells, stimulating augmentation of the immune response to foreign antigens from T lymphocytes. In the molecular mechanisms underlying MSU-induced inflammation, NLRP3 inflammasome following phagocytosis of MSU crystal was found to play a critical role in the gout-mediated innate immune response through production of active IL-1β and IL-18 [26].
Signal transduction through binding to ligands and cell surface receptors coordinates the innate immune response. Human mononuclear cells treated with MSU crystals alone did not show sufficient expression of IL-1β mRNA and protein compared to those administered a combined treatment of LPS and MSU crystals [58], which implies that the inflammatory response of gout requires to produce the synergic effect of secondary signal pathways other than those fostering MSU crystals. Among these cell surface receptors, TLRs such as TLR2 and TLR4 are involved in the production and secretion of inflammatory cytokines after exposure to uric acid in gouty inflammation [59]. MSU crystal-induced production of IL-1β and neutrophil influx were significantly suppressed in TLR2−/− and TLR4−/− mice models of gout. This indicates that TLR-dependent innate immunity in part through recognition of MSU crystals is responsible for acute gouty inflammation through direct interaction between TLRs and MSU crystals. However, there is a conflicting opinion regarding the essential role of TLRs in MSU crystal-induced inflammation. Chen et al. [60] found that MSU-induced inflammation was mediated by MyD88-dependent IL-1R but not MyD88-dependent TLR2 and TLR4, implying that IL-1β production and IL-1R signaling play essential roles in gouty inflammation. More recent studies suggest that TLRs indirectly coordinate gouty inflammation by recognizing ligands during the priming phase rather than direct ingestion of MSU. IL-1β production was markedly reduced in CD14−/− BMDMs to MSU crystals, whereas it was restored by coating with soluble CD14 but not with sTLR2 or sTLR4 [61]. Myeloid-related protein (MRP) 8 and MRP14 acting as endogenous TLR4 ligands were highly expressed in the serum and synovial tissue of gout patients, in MSU-stimulated human phagocytes, and in murine BMDMs treated with MSU and promoted MSU crystal-induced IL-1β secretion in a TLR4-dependent manner [62].
Several regulatory mechanisms have been found to be involved in NLRP3 inflammasome activation by MSU crystals such as mitochondrial ROS generation and K+ efflux, similar to NLRP3 inflammasome activation by other danger signals [9,63]. NLRP3-NEK7 complex formation downstream of potassium efflux by stimulation with MSU crystals, playing a role in oligomerization of ASC and activation of caspase-1, is an essential step to mediate NLRP3 inflammasome activation [48,49,64]. Recent studies demonstrated that MSU crystals triggered intracellular TXNIP transport from the nucleus to mitochondria, leading to NLRP3 inflammasome activation through interaction of NLRP3 and mitochondrial TXNIP [52,65]. The exact mechanism of diverse cytosolic mediators such as NEK7 and TXNIP required to activate the NLRP3 inflammasome under oxidative stress induced by MSU crystals has not been clearly determined.
3) IL-1β-medicated inflammatory cascades linked to inflammasome activation
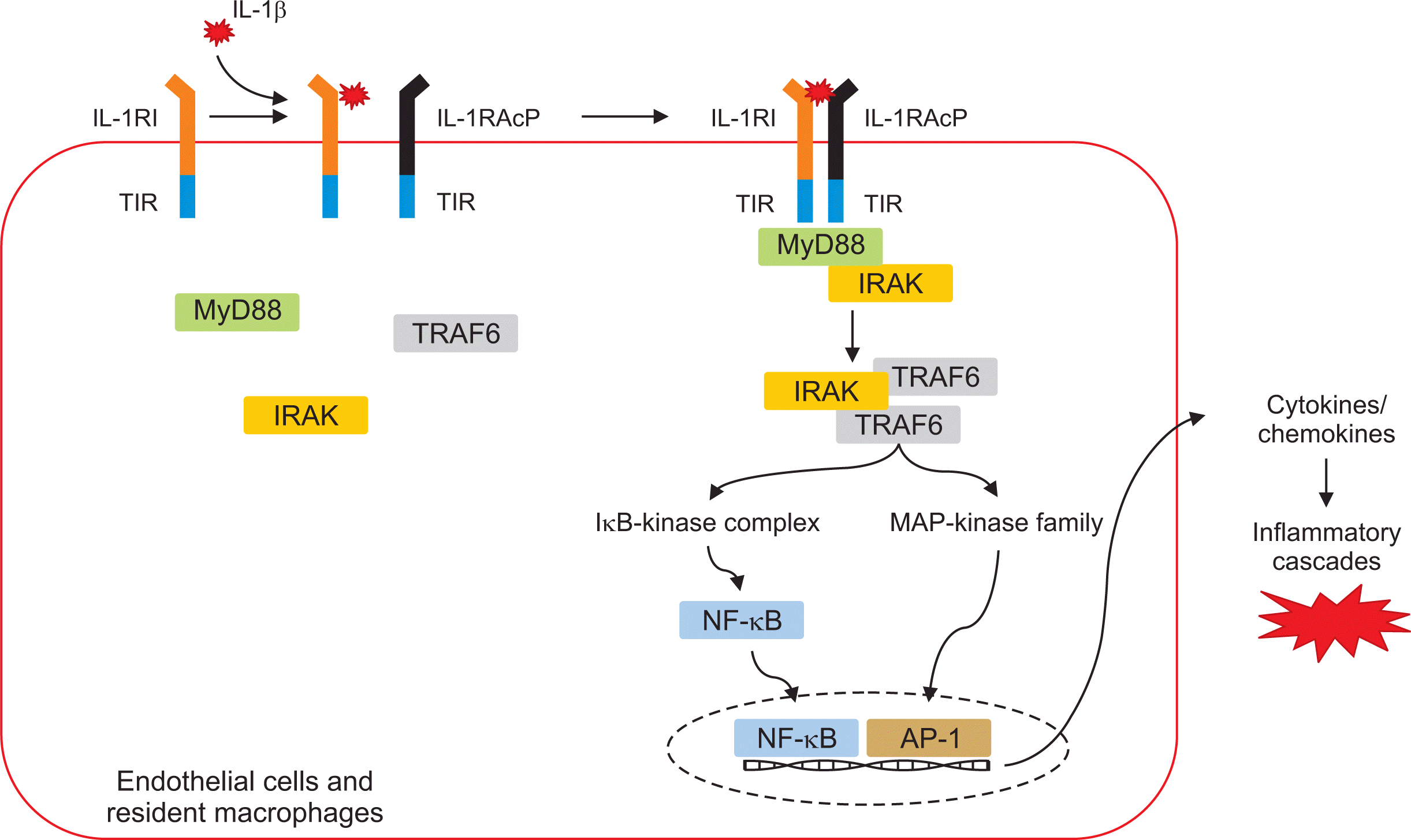
IL-1β is produced by innate immune cells such as monocytes, macrophages, and dendritic cells following binding to TLRs and danger signals and plays a role as a key signaling mediator to regulate both innate and adaptive immune systems [66]. The ternary complex structure of IL-1β:IL-1RI:IL-1RAcP allows initiation of the mechanism for IL-1 signal transduction (Figure 3). Cytokine binding to its receptor generally recruits some adaptor proteins such as MyD88, TRIF, and TRAM (TRIF-related adaptor molecule). Following binding to cytokine and its receptor, the Toll/IL-1 Receptor (TIR) domains that are located in the cytoplasmic portion of the two receptors initiate the inflammatory signaling cascade. Similarly, the binding of IL-1β to two receptors leads to dimerization of the cytoplasmic TIR domains of the two receptors and is accompanied by adaptors IRAK and MyD88 [67]. Recruitment of multiple adaptor and effector proteins such as IRAK and TNF receptor-associated factor-6 (TRAF6) activates proinflammatory transcription factors including NF-κB and mitogen-activated protein kinases (MAPKs). Sequentially, these intracellular signal pathways trigger the release of a variety of proinflammatory mediators such as cytokines, chemokines, and various enzymes.
The dysregulated IL-1 signaling pathway has been involved in numerous autoinflammatory diseases [68]. Gout is also considered an autoinflammatory disease induced by overexpression of IL-1β in the process of MSU crystal-mediated NLRP3 inflammation activation. IL-1β generated during the process of NLRP3 inflammasome activation triggers vasodilation of the endothelium at inflammatory sites and recruits IL-1R-expressing cells such as monocytes, resident macrophages, and neutrophils to arthritis joints through multiple chemokines including CXCL8/IL-8 and MCP-1 [63]. Several studies demonstrated that the IL-1β and IL-1R signal pathway is a master regulator for amplification of MSU crystal-induced inflammation in the pathogenesis of gout. MSU-induced neutrophil infiltration and expression of keratinocyte-derived chemokine and macrophage inflammatory protein-2 in the peritoneal cavity were significantly inhibited in MyD88−/− mice but not in TRAM−/−, Mal−/−, or TRIF−/− mice [60]. Mariotte et al. [69] found that resistance to expression of MSU-induced inflammatory cytokines such as IL-1β and IL-6 in MyD88 KO mice was markedly increased compared to that in IL-1RI KO mice. This suggests that adaptor MyD88 downstream of the IL-1β-medicated inflammatory cascade is required for gout inflammation.
Although caspase-1 of the NLRP3 inflammasome protein complex is central to producing the active form of IL-1β from precursor IL-1β in the pathogenesis in gout, NLRP3 inflammasome-independent IL-1β production and release via neutrophil-derived serine proteases have been identified in gout flares [70]. The Casp1−/− gout mice model induced by MSU crystal injection showed neutrophil influx and IL-1β protein in the peritoneal cavity, which was significantly attenuated by neutrophil elastase inhibitors [71]. Another study confirmed that alpha-1-anti-trypsin IgG1 Fc fusion protein (AAT-Fc) inhibited joint inflammation in a gout mouse model injected with MSU crystals and the fatty acid C16.0 by blocking neutrophil serine protease proteinase-3-mediated conversion of the pro-IL-1β into active IL-1β [72]. However, the exact mechanism by which these caspase-1-independent proteinases bypass the caspase-1 pathway and amplify the neutrophil-dominant inflammatory response in gout inflammation has not been elucidated.
Therapeutic targeting of NLRP3 inflammasome in gout
During the last 50 years, effective pharmacological management strategies against gout have consisted of anti-inflammatory therapy for acute gout attacks, urate-lowering therapy, and concomitant prophylactic treatment [1]. In particular, first-line anti-inflammatory drugs such as nonsteroidal anti-inflammatory drugs (NSAIDs), colchicine, and glucocorticoids have been used to control the acute inflammation of gout. According to a recent management guideline proposed by the American College of Rheumatology, IL-1 inhibitors are conditionally used in patients who show lack of effectiveness, poor tolerability, or contraindications to these anti-inflammatory drugs [1,73]. Furthermore, insight advances into the pathogenesis of gout provide evidence for novel therapeutic agents targeting NLRP3 inflammasomes and the IL-1β pathway.
1) Inhibitors for NLRP3 inflammasome activation
Significant efforts have been made to develop therapeutic agents that target the components and its substrates involved in NLRP3 inflammasome activation. Maturation of precursor IL-1β in the process of NLRP3 inflammasome is dependent on cysteine caspase-1 and serine proteases. In particular, caspase-1 is considered a core enzyme of NLRP3 inflammasome activation for production of IL-1β. Therefore, chemical compounds targeting caspase-1 have been developed to block NLRP3 inflammasome activation. VX-765, an orally absorbed prodrug of VRT-043198, is a potent and selective caspase-1 inhibitor [74]. VX-765 showed prophylactic efficacy for synovial inflammation and bone erosion in a collagen-induced arthritis model similar to that of rheumatoid arthritis [75]. However, there have been no clinical studies on the therapeutic effect of VX-765 for gout. The anti-inflammatory effect of human recombinant AAT-Fc fusion protein is derived from blockage of proteinase-3-mediated cleavage of the IL-1β precursor into active IL-1β [72]. Thus, AAT-Fc is considered a highly effective therapeutic option in reducing gouty arthritis. Blocking the assembly of NLRP3 inflammasome could be an important strategy for regulation of inflammation. MCC950 (also known as CP-456) is a small-molecule inhibitor of NLRP3 inflammasome through NLRP3-induced ASC oligomerization in human and mouse macrophages [76]. However, this drug specifically affects both canonical and noncanonical NLRP3 inflammasome and has no effect on AIM2, NLRP1, or NLRC4 inflammasome. MCC950 inhibited IL-1β release in LPS-primed BMDMs treated with other NLRP3 stimuli such as nigericin or MSU crystals [76,77]. It implicates that specific inhibitor of NLRP3 inflammasome MCC950 provides the possibility of manage numerous NLRP3-related inflammatory diseases such as gout.
Existing research has been applied to the treatment of gout inflammation with several chemical or natural medications used for other inflammatory diseases in clinical practice. Huang et al. [78] identified tranilast, an analog of a tryptophan metabolite and anti-allergic drug, to play a role as an NLRP3 inhibitor by blocking NLRP3-dependent IL-1β production in mouse models of gouty arthritis. A gastroprotective drug, rebamipide, inhibited ROS generation and conversion of pro-caspase-1 to active caspase-1 in human monocyte cell line THP-1 stimulated by MSU crystals, then resulting in suppression of IL-1β maturation [79]. Artemisinin, a well-known antimalarial agent with anti-inflammatory and immunoregulatory properties, inhibited NEK7 mRNA and protein expression in human macrophage U937 cells treated with MSU crystals, leading to downregulation of IL-1β expression and ultimate suppression of joint inflammation in a gout mouse model [64]. K+ efflux plays a central role in NLRP3 inflammasome activation following stimulation with danger signals [27,28]. In addition, artemisinin contributed to blockade of potassium efflux in human macrophage U937 cells under stimulation of MSU crystals with LPS [64]. Although these experimental medications might modulate the anti-inflammatory effect on gout inflammation at multiple signal pathways, medications targeting or modulating NLRP3 inflammasome activation are not available in clinical practice.
2) IL-1β/IL-1R inhibitors
Similar to anti-cytokine treatment for rheumatoid arthritis, evidence of clinical studies for blockade of IL-1 in acute gouty arthritis has been provided. The two IL-1 inhibitors anakinra and canakinumab showed therapeutic efficacy in patients with refractory gout (Table 3).
(1) Anakinra
Anakinra is a recombinant, non-glycosylated form of the human IL-1R antagonist that is approved by the US Food and Drug Administration (FDA) for treatment of rheumatoid arthritis. Regarding the therapeutic role of anakinra in gout, in a pilot, open-labeled study, 10 patients with refractory gout showed rapid and good response to anakinra without any adverse effects [80]. Ghosh et al. [81] assessed the effectiveness and safety of anakinra in 26 hospitalized patients with acute gouty arthritis and found that 73% experienced complete resolution of symptoms and signs of gout with no drug-related adverse effects. Another multicenter retrospective study of 40 gout patients found that most patients (90%) had a good response [82], although they identified seven infectious complications in patients with long-term use of anakinra (≥1 month). These findings suggest anakinra as an effective treatment for acute gouty arthritis refractory to first-line treatments. However, randomized controlled trials (RCTs) using anakinra in patients with acute gouty arthritis are needed to confirm these data.
(2) Canakinumab
Canakinumab is a human IL-1β monoclonal antibody that neutralizes inflammatory cytokine IL-1β. Significant clinical data have been provided on the therapeutic effect of canakinumab for acute gouty arthritis. Two 12-week multicenter double-blind RCT core studies with double-blind 12-week extensions including canakinumab 150 mg (n=230) or triamcinolone acetonide 40 mg (n=226) revealed that canakinumab significantly improved the 72-hour visual analogue scale pain score and physician-assessed tenderness and swelling compared to triamcinolone [83]. However, the canakinumab group had more frequent infectious adverse effects than the triamcinolone group, though most were of mild to moderate severity.
A double-blind RCT study in 432 patients with gouty arthritis evaluated whether canakinumab four times monthly prevented acute gout flares after initiating allopurinol compared to colchicine 0.5 mg daily for 16 weeks [84]. Patients treated with canakinumab showed a lower number of flares per patient and a shorter duration of flares compared to the colchicine group. This implies that canakinumab injection significantly reduced inflammatory symptoms and signs in acute gout, as well as the risk of gout flares. Canakinumab has provided sufficient evidence for a therapeutic impact on acute flares in gouty arthritis. Canakinumab was approved by the European Medicines Agency (EMA) in 2013 for patients with frequent gout attacks (≥3 attacks in the previous year) or who are refractory, intolerable, or contraindicated to first-line treatment. However, the FDA declined to approve the use of canakinumab in acute gout arthritis due to safety concerns regarding increased infection risk.
CONCLUSION
Activation of the innate immune system is initiated through PRRs such as NLRs, RLRs, and TLRs that recognize cellular and molecular pathogens and damages. Multi-protein platform NLRP3 inflammasomes recruit sensor NLRP3, the adaptor protein ASC, and inflammatory pro-caspase-1, leading to maturation of precursor pro-inflammatory cytokines such as IL-1β and IL-18 to the active forms of IL-1β and IL-18, which are mainly produced by innate immune cells. NLRP3 inflammasome activation is induced by a variety of intracellular and extracellular signaling mechanisms such as K+ efflux and Ca2+ mobilization, lysosomal destabilization, and ROS generation. Following NLRP3 inflammasome activation, IL-1 binding to IL-1RI and IL-1RAcP, referred to as the IL-1/IL-R complex, recruits diverse adaptors including MyD88, IRAK, and TRAF6, subsequently leading to activation of inflammatory transcription factors NF-κB and MAPKs. This augments the production of chemokines and proinflammatory cytokines that modulate the IL-1-mediated complex inflammatory cascade. However, despite extensive studies, the mechanisms of NLRP3 inflammasome activation and the IL-1 inflammatory cascade have not been clearly elucidated.
Evidence about complex intracellular signal components after uptake of MSU crystals, such as mitochondrial oxidative stress, TXNIP, and NEK7, suggests that they are indispensable for NLRP3 inflammasome activation in the pathogenesis of gout. Several therapeutic medication candidates have been developed to control the acute inflammation of gout by inhibiting NLRP3 inflammasome activation and blocking IL-1. In particular, IL-1 inhibitors such as anakinra and canakinumab showed potent therapeutic efficacy for acute symptoms of gout or prophylactic effects on gout flares. Despite much understanding of the pathogenesis of gout, there are many questions to be answered about the cellular and molecular mechanisms for development of new therapeutics. Future investigations into this ‘older disease’ will provide additional opportunities to control the inflammatory response.
 XML Download
XML Download