

 PDF
PDF Citation
Citation Print
Print


Introduction
Air pollution is one of the most concerning environmental risks to human society. The primary source of air pollutants is particulate matter (PM), which is categorized into three types according to particle size: coarse (<10 μm), fine (<2.5 μm), and ultrafine (<0.2 μm). The majority of particles contributing to airborne PM is ultrafine diesel exhaust particles (DEP) that are emitted from diesel engines (1).
DEP is a complex mixture of organic and inorganic material including polycyclic aromatic hydrocarbons (PAHs). There is increasing evidence to suggest that oxidative stress caused by PAH-induced reactive oxygen species (ROS) affects macrophages and epithelial cells and is the main driver of inflammation and damage in the lungs (2).
When alveolar macrophages and bronchial epithelial cells first encounter inhaled DEP, DEP trigger activation of macrophages to the M1 phenotype and release of pro-inflammatory cytokines (3). Furthermore, DEP activate mitogen-activated protein kinases (MAPKs) and nuclear factor-κB (NF-κB) to induce IL-6, IL-8, and GM-CSF, which are markers for pro-inflammation (4). Recent studies suggest that DEP also affects T cell diffe-rentiation. In a mouse model of multiple sclerosis, the aryl hydrocarbon receptor (AHR) regulates development of regulatory T cells (Treg) and T helper 17 (Th17) cells (5). Taken together, these findings indicate that DEP-induced PAHs enhance oxidative stress and activate pro-inflamma-tory responses in macrophages, bronchial epithelial cells, and naïve T cells.
Mesenchymal stem cells (MSCs) have been reported to modulate immune responses to tissue damage and inflammation by secreting numerous bioactive molecules, cytokines, growth factors, and chemokines and by modulating cell-to-cell contact (6). As a result of their hypoim-munogenic and immunosuppressive properties, MSCs show potential to modulate severe inflammatory responses (7). MSCs suppress proliferation of CD4+, CD8+, and pro-inflammatory type 1 helper T cells (Th1) to inhibit hyper-adaptive immune reactions (8). MSCs also inhibit pro-inflammatory M1 macrophages and activate anti-inflam-matory M2 macrophages (9). Previously, we have shown that the human MSC secretome, including transforming growth factor-beta (TGF-β), prostaglandin E2 (PGE2), indoleamine 2,3-dioxygenase (IDO), and extracellular vesicles (EVs), is responsible for the anti-inflammatory properties (10, 11). The mechanisms and therapeutic effects of MSCs in autoimmune and DEP-induced diseases have been studied; however, neither the nature of direct interactions between DEP and MSCs nor the precise mechanism of DEP effects on MSCs have been elucidated (12-14).
In this study, we investigated the detailed mechanisms of DEP effects on MSC stemness, immunomodulatory functions, and pro-inflammatory activation signal pathways.
Materials and Methods
Diesel exhaust particle preparation
The Standard Reference Material (SRM) 1650b diesel particulate matter was obtained from the National Institute of Standards and Technology (NIST, Gaithersburg, MD, USA). The DEP were suspended in dimethyl sulfoxide as a 10 mg/ml stock solution.
Human Wharton’s jelly-derived mesenchymal stem cells preparation and culture
Wharton’s jelly-derived mesenchymal stem cells (WJ-MSCs) were isolated from umbilical cords of healthy full-term babies as previously described (10). The procedures for tissue harvesting and obtaining informed consent were approved by the Asan Medical Center Institutional Review Board (Protocol no. 2015–3030). WJ-MSCs were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Tissue Culture Biologicals, Tulare, CA, USA), 1% Glutamax (Gibco), 1% antibiotic/antimycotic solution (Gibco), 25 ng/ml epidermal growth factor (EGF; Peprotech, Rocky Hill, NJ, USA), and 50 ng/ml basic fibroblast growth factor (bFGF; Peprotech) in a humidified atmosphere containing 5% CO2 at 37℃. Cells were passaged every 3∼4 days using 0.05% trypsin/EDTA. The cells were pretreated with N-acetyl-l-cysteine (NAC; 5 mM; Sigma-Aldrich, St. Louis, MO, USA), U0126 (10 μM; Cell Signaling Technology, Danvers, MA, USA), or parthenolide (PTL; 5 μM; Sigma-Aldrich) before stimulation with DEP (10 μg/ml) as indicated.
MTT assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) assay was performed to assess the effect of DEP on the viability of WJ-MSCs. WJ-MSCs were seeded into a 24-well plate at 5×104 cells/well with and without various concentrations of DEP for 48 hr. MTT reagent (0.5 mg/ml; Sigma-Aldrich) was added, and the cells were cultured for 3 hr at 37℃. The resulting formazan precipitate was solubilized in 0.04 M HCl, and the absorbance was measured at 570 nm using a SynergyTM H1 microplate reader (Biotek, Winooski, WT, USA).
Apoptosis assay
For apoptosis assessment, WJ-MSCs were seeded into a 96-well plate at 1×104 cells/well with DEP (0∼40 μg/ml) for 48 hr. The cells were stained with Annexin-V-FITC (BD Biosciences, Franklin Lakes, NJ, USA) and 7-AAD (BD Biosciences) following the manufacturer’s instructions and analyzed by flow cytometry.
Scratch wound healing assay
WJ-MSCs were seeded into a 24-well plate at 1.5×105 cells/well and cultured overnight to 100% confluency. The cells were incubated with 10 μg/ml mitomycin C (MMC; Sigma-Aldrich) for 1 hr to render them mitotically inactive. The cells were scratched with a 200 ul pipette tip and treated with 0∼10 μg/ml DEP. The cells migrating into the scratched area were photographed at 0, 18, and 36 hr, and migration distances were calculated using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
In vitro differentiation assay
WJ-MSCs were seeded into a 12-well plate at 5×104 cells/well and incubated with various concentrations of DEP for 48 hr. When the cells reached 70∼80% confluence, StemMACSTM Adipodiff Media and OsteoDiff Media (Miltenyi Biotec, Bergisch Gladbach, Germany) were added to assess adipogenic and osteogenic differentiation, respectively. The induction media were changed every three days. After three weeks, calcium deposits and lipid droplets were stained with Alizarin Red S (Sigma-Aldrich) and Oil Red O (Abcam, Cambridge, UK), respectively. For quantification, Alizarin-Red-S-stained cells were dissolved in 100 mM cetylpyridinium chloride (Sigma-Aldrich), and the absorbance was measured at 590 nm. Oil-Red-O-stained cells were dissolved in 100% isopropanol, and the absorbance was measured at 490 nm using a spectrophotometer.
RNA isolation and sequencing (RNA-seq)
RNA sequencing (RNA-seq) was performed by Bioneer Co. (Daejeon, Korea) using Illumina technology. Total RNA was extracted from the WJ-MSCs incubated with or without 5 μg/ml of DEP using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The mRNA sequencing library was generated using the Illumina TruSeq strand mRNA sample preparation kit (Illumina, San Diego, CA, USA) and sequenced using NovaSeq 6000 (2×150 paired end sequencing, Illumina) according to the manufacturer’s protocol. After removing the adapter sequence and filtering the low-quality readings using an in-house script, the filtered readings were mapped to the reference human genome using TopHat (15). For differential expression analysis, the gene expression of each group was quantified using cufflink v2.1.1 (16) by mapping the readings to the human gene annotation database (hg19). Next, the differentially expressed genes (DEGs) in the control and DEP-treated WJ-MSCs were identified based on absolute log2-fold change ≥1 and p<0.05 using Cuffdiff (17, 18). Then, heatmaps were generated using an in-house script, and clustering analysis was performed using a hierarchical clustering method. Volcano plots of DEGs were prepared using GraphPad Prism software (version 8.0.1; GraphPad Software, San Diego, CA, USA).
Gene ontology (GO) analysis and gene set enrichment analysis (GSEA)
We identified gene sets based on gene ontology (GO) into three categories: biological processes (BP), cellular components (CC), and molecular function (MF). The significance of the gene sets was calculated using gene set enrichment analysis (GSEA v3.0, https://www.gsea-msigdb.org/gsea/index.jsp). GO-based trend testing was performed using Fisher’s exact test.
Quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen), and 2 μg of total RNA was converted to cDNA using SuperscriptTM III reverse transcriptase (Invitrogen). Real-time PCR was performed using the SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) and measured using a MX3000P thermal cycler (Agilent Technologies, Santa Clara, CA, USA). β-actin was used as the reference gene for normalization. The primer sequences for qRT-PCR are listed in Supplementary Table S1.
Detection of intracellular reactive oxygen species (ROS)
Intracellular reactive oxygen species (ROS) were detec-ted using the peroxide-sensitive fluorophore 2’,7’-dichlorofluorescin diacetate (DCFDA, Sigma-Aldrich). WJ-MSCs were incubated with various concentrations of DEP for 48 hr. After incubation, cells were washed with PBS and incubated with 10 μM DCFDA in serum-free culture medium for 30 min at 37℃. The mean DCFDA fluorescence was analyzed using an Attune NxT flow cytometer (Thermo Fisher Scientific, Waltham, MA, USA).
Isolation of umbilical cord blood-derived mononuclear cells
Umbilical cord blood (UCB) samples were obtained from the Catholic Hematopoietic Stem Cell Bank (CHSCB) under approval of Institutional Review Board (IRB) of the Seoul National University (IRB No. E2011/001-010). Human mononuclear cells (MNCs) were isolated by Ficoll gradient centrifugation as previously described (10), and were cultured in RPMI 1640 medium (Gibco) supplemented with 10% FBS.
T cell proliferation assay
WJ-MSCs were pre-cultured with or without NAC for 1 hr, and then various concentrations of DEP were added for 48 hr. Subsequently, the WJ-MSCs were treated with 10 μg/ml MMC for 1 hr to arrest cell proliferation, and the cells were seeded at 1×104 cells per well in a 96-well plate. After 24 hr, MNCs were labeled with 2 μM 5,6-carboxyfluorescein succinimidyl ester (CFSE; Thermo Fisher Scientific), and 1×105 MNCs were added to each well containing WJ-MSCs, in the presence of anti-CD3/CD28 microbeads (Gibco) and recombinant human IL-2 (30 U/ml; PeproTech). After six days of incubation, the cells were stained with fluorescence-labeled human monoclonal antibodies against CD45-APC-H7, CD3-BV510, CD4-APC, and CD8-BV421 (BD Biosciences). Proliferation of total T cells and T cell subpopulations was measured by dilution of CFSE using a FACScanto flow cytometer (BD Biosciences). Viable lymphocytes were gated on 7AAD-negative cells.
WJ-MSCs small interfering RNAs (siRNA) transfection
To silence the expression of cFos, WJ-MSCs were transiently transfected with specific siRNAs (Bioneer) using the LipofectamineTM RNAiMAX reagent (Invitrogen) according to the manufacturer’s protocol. After 4 hr of transfection, 10 μg/ml of DEP was added, and the cells were incubated at 37℃ for 24 hr.
Western blot analysis
The cells were lysed using RIPA buffer (Thermo Fisher Scientific), and the protein concentrations were determined using a BCA assay kit (Thermo Fisher Scientific). Equal amounts of proteins were obtained after SDS-PAGE separation and analyzed using primary antibodies against cFos, ERK, p-ERK, and p-IκBα (Cell Signaling Technology), and GAPDH (Santa Cruz Biotechnology, Dallas, TX, USA). The bands were visualized using an enhanced chemiluminescence assay kit (Thermo Fisher Scientific) and luminescent image analyzer (LAS-3000 system; Fujifilm, Tokyo, Japan). For quantification, ImageJ software (National Institutes of Health) were used to analyzed the bands intensity.
Dextran sulfate sodium (DSS)-induced colitis mice
All animal experiments were carried out in accordance with the approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul National University (protocol no. SNU-201021-2-1). Colitis was induced by administration of 3% dextran sulfate sodium (DSS; MP Biomedicals, Santa Ana, CA, USA) in drinking water for 7 days. 8-week-old C57BL/6 mice were randomly assigned to 5 groups (n=10 per group: (1) Negative control, (2) DSS only, (3) DSS with WJ-MSC, (4) DSS with DEP-treated WJ-MSC (DEP group), (5) DSS with NAC pretreated DEP-treated WJ-MSC (NAC group). Mice were injected intraperitoneally with PBS or cells (2×106 cells in 200 μl) on day 1 after DSS drinking. The severity of colitis was assessed daily using the disease activity index (DAI). All the mice were sacrificed on day 12 and their colon lengths and weights were measured. Myeloperoxidase (MPO) activity assay and histopathological evaluation were performed as previously described (11).
Statistical analysis
For statistical analysis, all experiments were performed at least in triplicate. Where data were normally distributed, the significance was determined using Student’s t-test or a one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. The data are presented as the mean±standard deviation. All statistical analyses were performed using GraphPad Prism software (version 8.0.1).
Results
Diesel exhaust particles (DEP) impair WJ-MSC functions
Many studies have shown that DEP activate pro-infla-mmatory responses and affect various cellular functions in diverse cell types (19-22). We therefore investigated the effects of DEP on stemness and immunomodulatory functions of WJ-MSCs. First, we assessed DEP-induced toxicity using the MTT assay; incubation with 10 μg/ml or lower concentrations of DEP did not affect the cell viability, but the viability was significantly reduced at 20 μg/ml and 40 μg/ml DEP (Fig. 1a). Moreover, higher concentrations of DEP significantly induced early and late apoptosis (Fig. 1b). Based on these results, non-apoptotic concentrations of DEP (0∼10 μg/ml) were used in subsequent experiments.
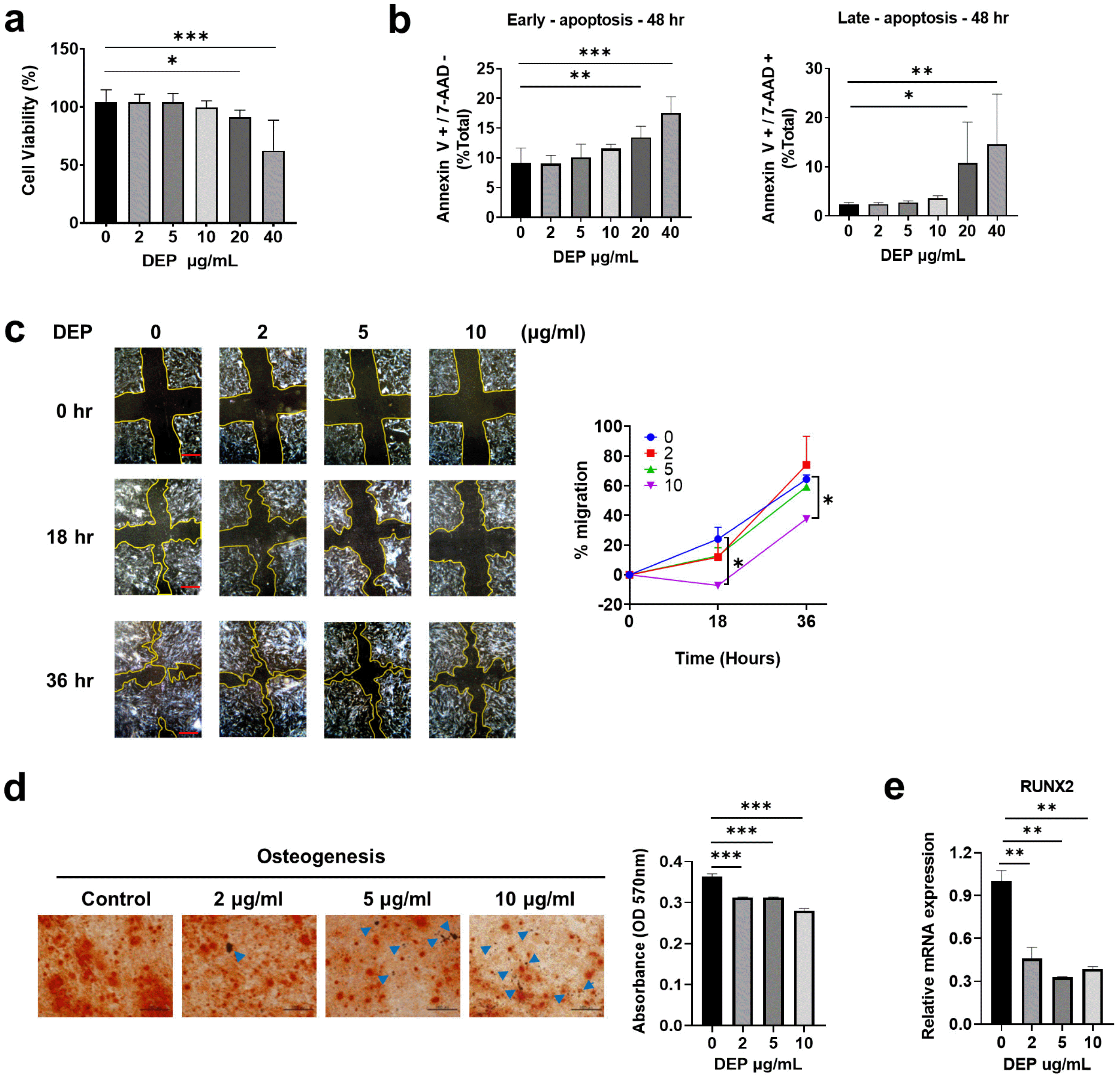
Fig. 1
Diesel exhaust particles (DEP) affect WJ-MSC function. (a, b) The WJ-MSCs were treated with DEP (0∼40 μg/ml) for 48 hr. (a) Cell viability was assessed using an MTT assay. (b) Early and late apoptosis were measured by labeling with Annexin-V-FITC and 7-AAD for flow cytometry analysis. (c) A scratched wound healing assay of DEP-treated WJ-MSCs (0∼10 μg/ml) was performed to analyze cell migration into the scratched area. After 18 and 36 hr DEP treatment, migration into the scratched area was measured. (Scale bar: 200 μm). (d, e) DEP-treated WJ-MSCs were cultured with osteogenic differentiation medium for 21 days. (d) Representative light microscopic images of osteocytes. Calcium deposits in cells cultured under osteogenesis were stained with Alizarin Red S (scale bar: 100 μm) and quantified by measuring absorbance using a spectrophotometer. Blue arrows indicate DEP. (e) Expression of an osteogenic gene (RUNX2) was quantified by qRT-PCR with β-actin as the reference gene. Data are presented as the mean±S.D. of triplicate experiments (*p<0.05; **p<0.01; ***p<0.001).
![]()
To test the migration and differentiation potential of DEP-treated WJ-MSCs, a scratch wound assay was performed, and in vitro differentiation (adipogenesis and osteogenesis) was assessed. The cell-based wound healing assay showed that migration of WJ-MSCs incubated with 10 μg/ml DEP was significantly reduced compared with that of control WJ-MSCs (Fig. 1c). When cultured under osteogenesis differentiation conditions, calcium deposition was reduced by incubation with DEP in a concentration-de-pendent manner (Fig. 1d), as was the expression of the osteogenic differentiation marker RUNX2 (Fig. 1e). However, there were no significant effects on adipogenic differentiation at any DEP concentration tested (Supplementary Fig. 1a and 1b). These results indicate that DEP inhibits migration and osteogenic differentiation of WJ-MSCs.
DEP increases ROS production and pro-inflammatory cytokine expression in WJ-MSCs
RNA sequencing (RNA-seq) was performed to examine changes in the gene expression profile of DEP-treated WJ-MSCs. Global differences in gene expression profiles were visualized by the hierarchical clustering heat map and the volcano plot. Clustering analyses showed 504 differentially expressed genes (DEG) in control MSCs and DEP-treated WJ-MSCs (Fig. 2a). These genes were used in gene ontology (GO)-term analyses to identify overrepresented biological functions. The enriched GO-categories identified in DEP-treated WJ-MSCs included cellular processes, metabolic processes, and biological regulation (Supplementary Fig. 2a and 2b). Among the most upregulated genes related to metabolic processes in DEP-treated WJ-MSCs, we focused on cFos, as it provides a possible link to DEP-induced pathology (Fig. 2b). We confirmed that cFos mRNA expression was significantly increased after incubation of WJ-MSCs with DEP (Fig. 2c).
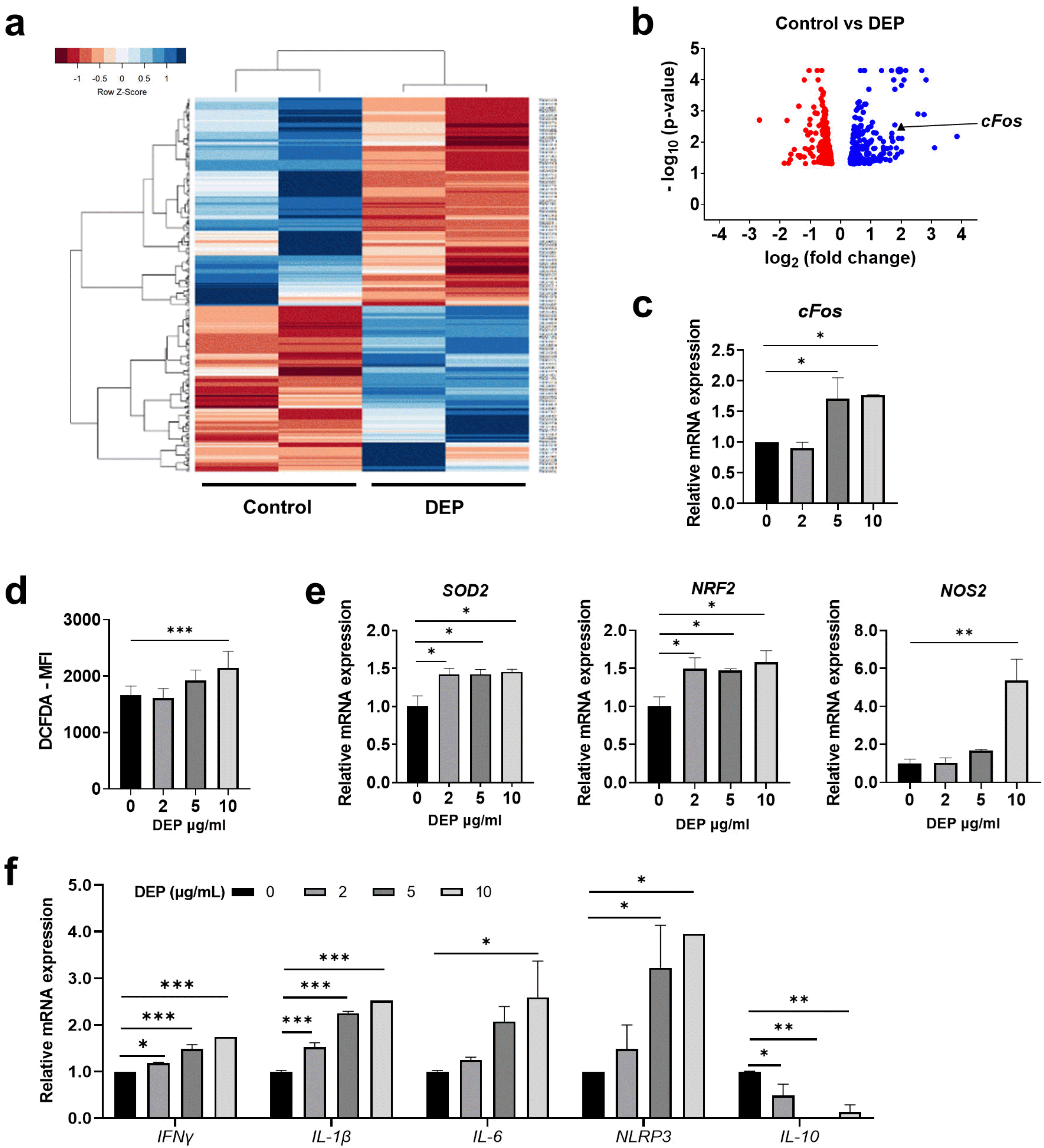
Fig. 2
DEP-treated WJ-MSCs express ROS and pro-inflammatory genes. (a) Heatmap of hierarchical clustering. Differentially expressed gene (DEG) profile for the control and DEP-treated WJ-MSCs were analyzed (p-value<0.05). (b) Volcano plot highlighting DEGs for the control and DEP-treated WJ-MSCs and notable genes of interest. The blue dots represent significantly upregulated genes, and the red dots represent significantly downregulated genes (|log2 FC|≥1 and FDR<0.01). (c) Transcription of cFos was quantified by qRT-PCR. (d) The WJ-MSCs were incubated with DEP at the indicated concentrations for 6 hr. Intracellular ROS levels were measured using the ROS-sensitive fluorophore 2’,7’-dichlorofluorescin diacetate (DCFDA) and analyzed by flow cytometry. (e) Transcription of ROS-related genes was determined by qRT-PCR and normalized to β-actin expression. (f) Transcription of inflammation-related genes was determined by qRT-PCR and normalized to β-actin expression. The data are presented as the mean±S.D. of three independent experiments (*p<0.05; **p<0.01; ***p<0.001).
![]()
As DEP-induced pathology is largely due to ROS-mediated cellular toxicity, we assessed induction of ROS in DEP-treated WJ-MSCs using DCFDA staining. At 10 μg/ml DEP, intracellular ROS increased significantly (Fig. 2d), and expression of ROS-related genes SOD2, Nrf2, and NOS2 was increased (Fig. 2e). We next investigated the regulation of inflammatory genes by DEP in WJ-MSCs. Expression of pro-inflammatory genes IFNγ, IL-1β, IL-6, and NLRP3 was significantly upregulated by DEP treatment, and the anti-inflammatory cytokine IL-10 was downregulated (Fig. 2f). These results indicate that DEP upregulates ROS-related genes and pro-inflammatory genes in WJ-MSCs.
DEP inhibits immunomodulatory properties of WJ-MSCs via ROS
To determine whether DEP-induced ROS are implicated in the immunosuppressive properties of WJ-MSCs, we performed a T cell proliferation assay in the presence of DEP and an ROS inhibitor (N-acetyl-l-cysteine, NAC). DEP-treated WJ-MSCs with or without added NAC were co-cultured with CFSE-labeled human umbilical cord blood-derived mononuclear cells that were activated with CD3/CD28 Dynabeads and IL-2. DEP inhibited WJ-MSC-mediated reduction of the proliferation of total lymphocytes and of CD4+ and CD8+ T lymphocytes in a concentration-dependent manner. This inhibitory effect was significantly attenuated by the ROS inhibitor (Fig. 3a and 3b, Supplementary Fig. 3a). The percentage of MNCs per cell division shifted to later cycles (cycle 4, 5) depending on the concentration of DEP and returned to the previous cycle (cycle 2, 3) when the WJ-MSCs were incubated with NAC (Fig. 3c, Supplementary Fig. 3b). Taken together, these data indicate that DEP-induced ROS are a major inhibitor of the immunosuppressive effects of WJ-MSCs.
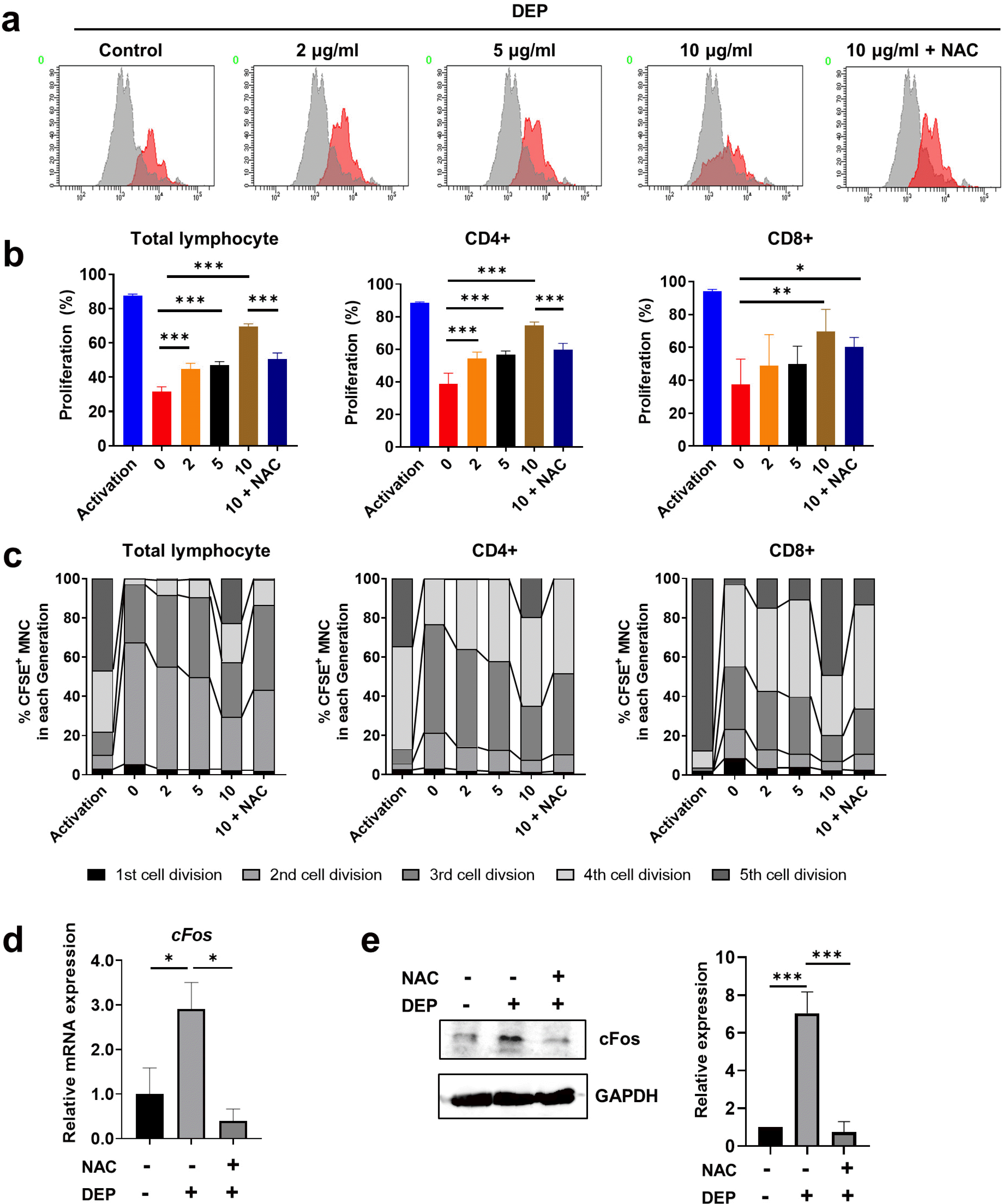
Fig. 3
DEP-induced ROS inhibits the immunomodulatory effects of WJ-MSCs. (a∼c) WJ-MSCs were pretreated for 30 min with NAC (5 mM) and then were mitotically inactivated after treating with various concentrations of DEP for 48 hr. Cells were co-cultured with CFSE-labeled human mononuclear cells (MNCs) that were activated by CD3/CD28 Dynabeads and IL-2. After six days of co-culture, the percentage of T cell proliferation was measured using flow cytometry. (a) Representative histogram of total lymphocyte proliferation. (b) Proliferation of total, CD4+ T, and CD8+ T lymphocytes was quantified. (c) The number of cell divisions of total, CD4+ T, and CD8+ T lymphocytes was quantified based on dilution of CFSE. (d, e) Cells were pretreated for 30 min with NAC and then were stimulated with 10 μg/ml DEP for 24 hr. (d) cFos mRNA was determined by qRT-PCR and normalized to β-actin mRNA. (e) cFos expression was quantified by western blot analysis. The data represent results from three independent experiments and are presented as mean±SD (*p<0.05; **p<0.01; ***p<0.001).
![]()
cFos is involved in inhibition of immunomodulatory function of DEP-treated WJ-MSCs
A previous study showed that cFos is a direct downstream target of ROS and is considered an ROS-induced pro-inflammatory gene (23). Therefore, we investigated whether ROS are implicated in DEP-induced cFos in WJ-MSCs. ROS inhibitor treatment significantly inhibited DEP-induced cFos mRNA and protein expression in WJ-MSCs (Fig. 3d and 3e). To investigate involvement of cFos in the immunomodulatory effects of WJ-MSCs, WJ-MSCs were transfected with specific siRNAs against cFos and cultured in the presence of 10 μg/ml of DEP. We confirmed siRNA-mediated knockdown of cFos at both the mRNA and protein levels in DEP-treated WJ-MSCs (Fig. 4a and 4b). Knockdown of cFos reversed the increase in T cell proliferation in DEP-treated WJ-MSCs (Fig. 4c and 4d, Supplementary Fig. 4a). As indicated in Fig. 4e, the percentages of MNCs per cell division in cFos-knockdown WJ-MSCs increased in the 2nd and 3rd division cycles compared with DEP-treated WJ-MSCs (Fig. 4e, Supplementary Fig. 4b). In DEP-treated cFos-depleted WJ-MSCs, the osteogenic potential was markedly greater than in DEP-treated control WJ-MSCs (Fig. 4f). In addition, up-regulated pro-inflammatory cytokines, such as IL-1β, IL-6, and NLRP3, in DEP-treated WJ-MSCs were decreased after cFos depletion whereas the anti-inflammatory cytokine IL-10 was increased (Fig. 4g). These results indicate that DEP inhibits the immunomodulatory function of WJ-MSCs through cFos signaling.
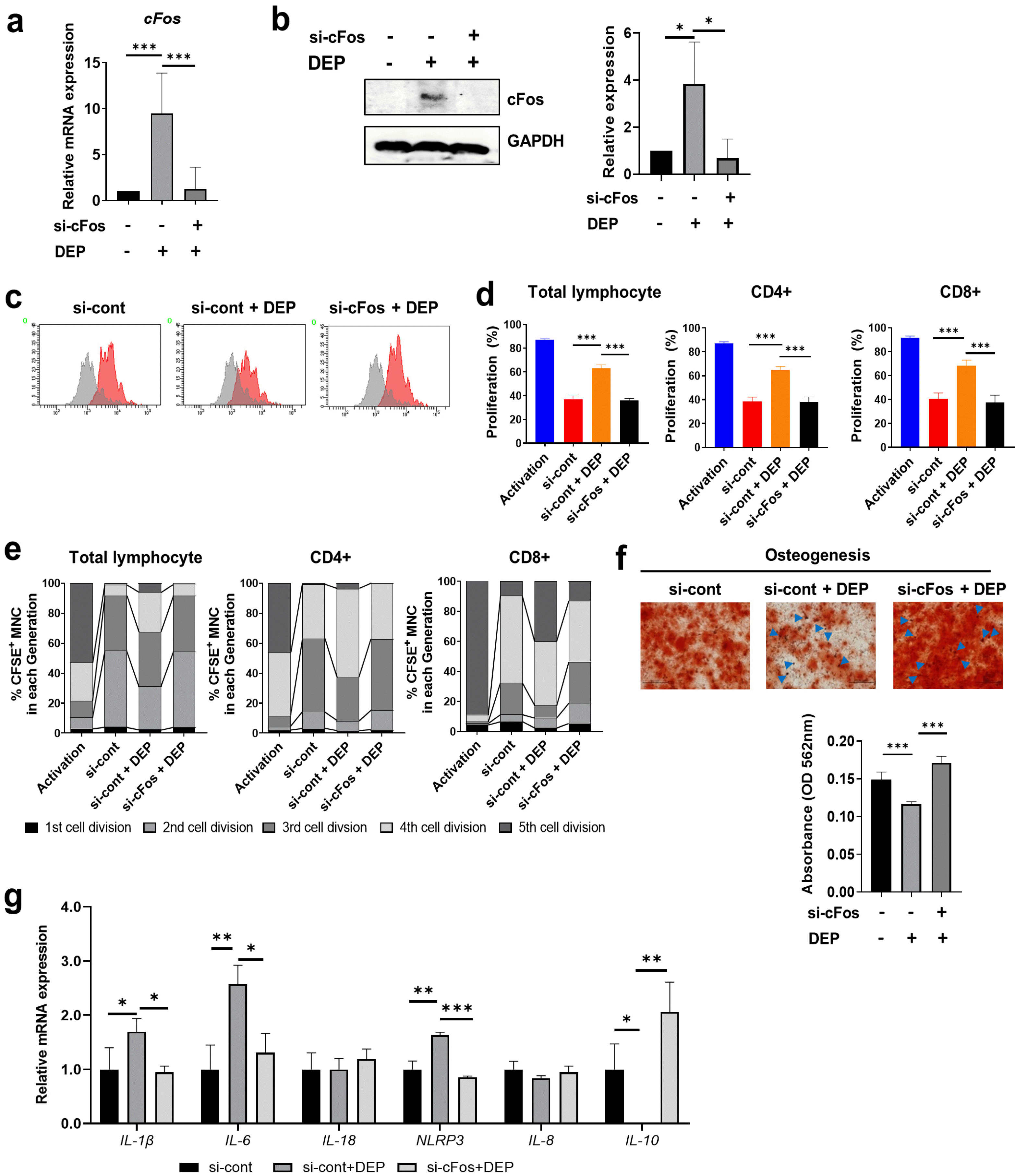
Fig. 4
DEP-induced cFos expression inhibits the immunomodulatory function of WJ-MSCs. WJ-MSCs were transfected with control siRNA (si-cont) or cFos siRNA (si-cFos) for 4 hr and then were stimulated with 10 μg/ml DEP. After 12 hr, knockdown of cFos was confirmed by qRT-PCR (a) and western blot (b). (c∼e) After 48 hr of incubation, cells were mitotically inactivated and co-cultured with MNCs as previously described. (c) Representative histogram of total lymphocyte proliferation. (d) Proliferation of total, CD4+ T, and CD8+ T lymphocytes was quantified. (e) Number of cell divisions of total, CD4+ T, and CD8+ T lymphocytes was quantified based on dilution of CFSE. (f) si-cFos-transfected WJ-MSCs were stimulated with DEP for 24 hr and then cultured with osteogenic differentiation medium for 14 days. Calcium deposits were stained with Alizarin Red S. Light microscopy images of osteocytes show representative images from three independent experiments (Scale bar: 100 μm). Absorbance of calcium deposits was measured using a spectrophotometer. (g) Transcription of IL-1β, IL-6, IL-8, IL-18, and NLRP3 was determined by qRT-PCR and normalized to β-actin transcription. Data are presented as mean±S.D. of triplicate experiments (*p<0.05; **p<0.01; ***p<0.001).
![]()
DEP activates cFos through ERK signaling
Increased concentrations of proinflammatory cytokines in human airway epithelial cells after DEP exposure have been reported to be associated with activation of NF-κB, cFos, and MAPK (24). Similarly, DEP increased phosphorylation of ERK and IκBα in a concentration-de-pendent manner in WJ-MSCs (Fig. 5a). To examine whether ERK and NF-κB activation is involved in DEP-induced cFos signaling, WJ-MSCs were pre-treated with the ERK kinase inhibitor U0126 and the NF-κB inhibitor parthenolide (PTL). The U0126 and PTL almost completely blocked the activation of ERK and NF-κB, respectively (Fig. 5b), and U0126 but not PTL significantly inhibited DEP-induced cFos expression (Fig. 5c). Taken together, these results indicate that inhibition of the immunomodulatory function of WJ-MSCs by DEP is mediated through ROS/ERK/cFos signaling.
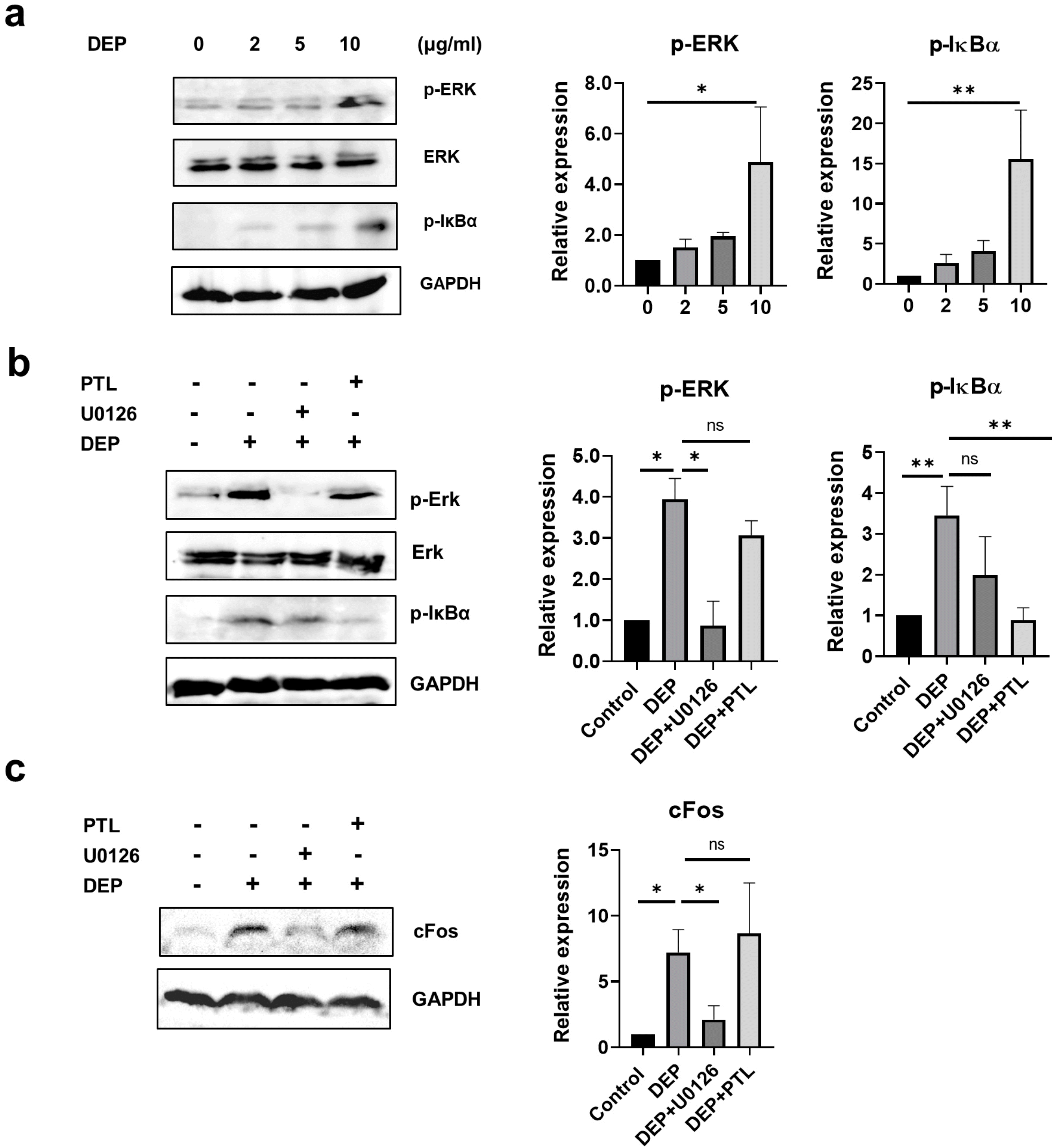
Fig. 5
DEP regulates cFos through ERK signaling. (a) WJ-MSCs were treated with DEP (0∼10 μg/ml) for 1 hr. Activation of p-ERK and p-IκBα was determined by western blot analysis. (b, c) WJ-MSCs were pretreated with U0126 (10 μM) or PTL (5 μM) and then were treated with 10 μg/ml of DEP for 1 hr. Expression of p-ERK, p-IκBα, and cFos was analyzed and quantified by western blotting (ns>0.05; *p<0.05; **p<0.01).
![]()
DEP-induced ROS impairs the protective effect of WJ-MSCs against DSS-induced colitis in mice
To evaluate whether DEP-induced ROS affected the therapeutic effect of WJ-MSCs in DSS-induced colitis, WJ-MSCs cultured with DEP and NAC were administered to mice 1 day after colitis induction (Fig 6a). As shown in Fig. 6a and 6b, mice in the DEP group displayed continuous body weight loss and remarkably elevated disease activity index (DAI). However, the NAC group was significantly ameliorated body weight loss and DAI as well as increased the survival rate (Fig. 6a∼c). Simultaneously, the colons of the DEP group were obviously shortened when compared to the WJ-MSC group whereas NAC group significantly increased the colon length and decreased the neutrophil infiltration (Fig. 6d and 6e). Histological examination showed that NAC group significantly rescued the submucosal thickening, the destruction of the epithelium, infiltration of inflammatory cells (Fig. 6f). The NAC group also had reduced the histopathological scores compared with the DEP group (Fig. 6g). Expression of pro‐inflammatory cytokines, including IFN‐γ and TNFα was decreased in colon tissue of NAC group compared to DEP group (Fig. 6h). Th1, Th17 and Treg, known to play an important role in the pathogenesis of inflammatory bowel disease, were analyzed in the colon of mice with experimental colitis. The DEP group significantly increased the expression levels of Th1 and Th17 lineage transcription factors compared to the WJ-MSC group, whereas the NAC group significantly decreased the expression levels of Th1 and Th17 transcription factors (Fig. 6i). However, Treg did not show any significant difference among groups. Taken together, these data indicate that DEP-induced ROS is involved in inhibiting the therapeutic effect of WJ-MSCs in DSS-induced colitis.

Fig. 6
DEP-induced ROS impairs the therapeutic effect of WJ-MSCs in colitis model. (a) The percentage of body weight change during entire experiment. (b) DAI scores were measured on day 7. (c) The survival rate of mice. (d) Mice were sacrificed on day 12, the length of colons was measured. (e) MPO activity was measured on day 12. (f) Colon tissue were examined using H&E staining and (g) histological scores were shown. The scale bars represent 500 μm and 200 μm. Expression levels of inflammatory cytokines (h) and Th1, Th17 and Treg associated cytokines (i) in colonic tissues were analyzed by qRT‐PCR (n=3 mice per group; *p<0.05; **p<0.01; ***p<0.001).
![]()
Discussion
Epidemiological studies have suggested that DEP exposure is associated with increased cardiovascular and respiratory morbidity and mortality. It has been suggested that DEP induce not only lung inflammation but also systemic damage of our body as lung inflammation possibly leak over to the cardiovascular system. Airborne PM were associated with dermatological, ocular, respiratory, cardiovascular, neurological, immunological, metabolic diseases and cancer (22). Inhaled DEP possibly penetrate into the alveolar system; DEP binds to the protein transport system in pulmonary epithelial cells, passes into the circulatory system, and is transported to secondary target organs, such as the heart and liver, where it causes inflammation (25). Previous studies have shown that DEP induces ROS generation in endothelial cells exposed to high concentrations of DEP, which leads to cellular apoptosis with Mdm2 depletion (26). In addition, WJ-MSCs might modulate endothelial responses such as leukocyte recruitment or vascular inflammatory responses (27). Although DEP has been reported to induce inflammation and apoptosis of human bone marrow (BM)-derived MSCs (22), the exact mechanism of the interactions between DEP and WJ-MSCs has not been elucidated. In light of previous studies indicating that PAHs in DEP enhance oxidative stress and activate pro-inflammatory responses in macrophages, bronchial epithelial cells, and naïve T cells, we evaluated the direct effect of DEP on the immunomodulatory function of WJ-MSCs (19, 23, 28).
Activation of NF-κB and expression of cFos by DEP exposure have been reported to contribute to the proliferation and pro-inflammatory processes of human bronchial epithelial cells (23). In the present study, we used RNA sequencing to identify 504 genes with markedly altered expressions in DEP-treated WJ-MSCs, and the most enriched gene set was that for metabolic processes, including cFos. DEP-induced effects in the lungs have been reported to involve cFos, but the possible relationship between regulation of pro-inflammatory mediators and cFos has not been thoroughly investigated (29). We demonstrated that DEPs induced pronounced expression of cFos and significantly increased the expression of pro-inflammatory factors such as IFNγ, IL-1β, IL-6, and NLRP3. Our findings suggest that cFos plays an important role in activation of DEP-induced NLRP3 inflammasome signals, since the expression of pro-inflammatory cytokines was almost abolished by inhibition of cFos.
Indeed, DEP has been reported to induce ROS, which are involved in cytokine formation, cytotoxicity, and DNA damage (1, 22). In addition, ROS can reduce the immunomodulatory functions of WJ-MSCs and promote their senescence and inflammation (30). In the present study, we confirmed that DEP increases ROS production in WJ-MSCs. In addition, DEP exposure inhibited the immunosuppressive effect of WJ-MSCs on T cell proliferation, and this response was abolished by pretreatment with the anti-oxidant NAC. Furthermore, WJ-MSCs incubated with DEP impaired the therapeutic effect in the experimental colitis mice and increased Th1 and Th17 cell responses, which were also abolished by the antioxidant NAC pretreatment. Our results indicate that DEP inhibits the immunosuppressive effects of WJ-MSCs by modulating ROS production. Although the exact mechanism of DEP-induced immune cell activation has not been studied thoroughly, it has been suggested that DEP increase bronchial hypersensitivity in asthmatic patients by enhancing basal T cell activation (2, 31). In the current study, we found that DEP did not directly affect T cell proliferation (Supplementary Fig. 3c) but inhibited WJ-MSC-mediated suppression of T cell proliferation. The antioxidant NAC inhibited the DEP-induced increase in cFos expression, suggesting that stimulation of cFos likely is dependent on activation of ROS. In addition, cFos inhibits osteogenesis and adipogenesis in immortalized human MSC progenitor cells (32). Our findings suggest that cFos activation is important for DEP inhibition of the osteogenic differentiation of WJ-MSCs because inhibition of their differentiation was reversed by cFos inhibition.
Exposure to DEP and the consequent oxidative stress have been shown to activate redox-sensitive transcription factors and various signal transduction pathways, including AP-1, NF-κB, MAPKs, p38, and JNK in airway epithelial cells (24). Activation of these proteins promotes the transcription of proinflammatory mediators, triggering the characteristic pulmonary inflammatory response of DEP exposure (33). We detected DEP-induced activation of p-IκBa and p-ERK; however, DEP-induced cFos expression was only reduced by ERK inhibition and was not affected by NF-κB inhibition. Similarly, DEP can induce release of pro-inflammatory cytokines through NF-κB or AP-1 regulatory pathways (34). Although no direct immunomo-dulatory function analysis of NF-κB was performed in this study, NF-κB activation induced by DEP could be attributed to stimulation of immune system responses; however, further study is needed to address this hypo-thesis.
It has recently been reported that atmospheric PM causes inflammation in lung cells, and that exposure to PM can increase the sensitivity and severity of symptoms in COVID-19 patients (35). Furthermore, air pollutants induce angiotensin converting enzyme 2 (ACE2) receptor expression in human epithelial cells (36, 37). Because WJ-MSCs have the potential to suppress immune system hyperactivity and inhibit cytokine storms with low expression of ACE2, COVID-19 patients were administered WJ-MSCs in a recent COVID-19 clinical trial (38, 39). Our study showed that DEP significantly impaired the immunomodulatory properties of WJ-MSCs; future studies of DEP-ACE2 interactions can inform potential therapeutic approaches for COVID-19 based upon WJ-MSCs.
Here we provide evidence that exposure to DEP enhances the expression of pro-inflammatory cytokines and suppresses the immunomodulatory properties through mechanisms involving ROS/ERK/cFos pathway in WJ-MSCs. In addition, we confirmed that DEP-induced ROS damage impairs the therapeutic efficacy of WJ-MSCs against inflammatory bowel disease, using DSS-induced colitis model. Therefore, exposure to DEP can affect the regenerative and immunomodulatory properties of WJ-MSCs and influence the pathophysiology of DEP-related diseases. Modulation of ROS/ERK/cFos signaling pathways in WJ-MSCs could be a novel target for acute and chronic inflammatory processes induced by DEP exposure.
Supplementary Materials
Supplementary data including one table and four figures can be found with this article online at https://doi.org/10.15283/ijsc21178.
 XML Download
XML Download