

 PDF
PDF Citation
Citation Print
Print


Introduction
Hypoxic-ischemic brain injury (HIBI) is a severe brain disease caused by oxygen deprivation and limited blood flow to the brain. It could cause irreversible necrosis, cell apoptosis, and neuron death, resulting in high mortality and profound long-term neurologic disability in survivors (1-3). Most return of function after HIBI occurs early, while behavioral recovery tends to plateau during chronic phase revealing the likely scale of permanent disability (4). Current treatment of HIBI is focused on optimizing the balance between cerebral oxygen supply and consume to reduce secondary injury (5). Despite numerous studies aimed at improving the outcomes of HIBI, no significant improvement has been observed over the past decades (5, 6). Neural stem cells (NSCs) remain in the mammalian brain throughout life, they are self-renewing, multipotent cells that generate the neurons and glia (7). Previous studies showed that NSCs can be activated by central nervous system (CNS) injury and participates in the restoration process of nerve injury (8). It provides a novel and promising therapeutic strategy for CNS injury associated with HIBI. However, it is reported that the infarct transitions from the acute to the chronic phase might lead to a glial scar with abnormal extracellular matrix (ECM), activated microglia/macrophages and abnormal blood supply (4, 9, 10). Ischemic injury could also induce excessive release of inflammatory cytokines, overproduction of reactive oxygen species (ROS) and other cascades of interactive events in the microenvironment (11, 12). All these factors can upset the fragile balance of NSC niche, affect the renewal and differentiation of NSCs and finally induce cell death. Although transplantation of NSCs is a promising approa-ch for treating ischemic injury, the hash microenvironment with excessive inflammatory cytokines and ROS largely limits the capacity of NSCs to repair the injured brain area. Hence, protecting NSCs against HIBI-induced oxidative stress and inflammation and promoting NSC survival in the harsh surrounding microenvironment may contribute to the clinical application of NSCs transplanta-tion.
Bone morphogenetic proteins (BMPs) are secreted hetero- or homodimeric proteins belonging to the transfor-ming growth factor-β (TGF-β) superfamily. It’s reported that BMPs are expressed in the central nervous system and involved in the pathologies of ischemic brain injury, in addition to their physiological roles in embryonic and adult neurogenesis (13). Feng and Hu (14) demonstrated that BMP-9 pretreatment may prevent cerebral ischemic-reperfusion injuries by alleviating neuronal apoptosis and promoting neuron growth. In neonatal mouse brain with hypoxia-ischemia, BMP-4 expression was increased, while the BMP antagonist noggin protects the white matter against ischemic damage (15). Lee et al. (16) found that BMP-7 could promote neuronal survival and growth after oxygen and glucose deprivation (OGD) injury. BMP-6, a member of the BMP subfamily, has been identified as a multifunctional cytokine capable of regulating cell proliferation, differentiation and apoptosis of multiple cell types, including mesenchymal stem cells (17), hematopoie-tic stem cells (18) and neural stem cells (19). Previous studies reported that BMP-6 could block low potassium-mediated apoptosis in cultured cerebellar granule cells (20), inhibit oxidative stress-induced injury in retinal pigment epithelial cell (21) and protect fibroblast-like synoviocytes against nitric oxide-induced apoptosis (22). However, the effects of BMP-6 on NSC survival in HIBI has not been fully elucidated.
In this study, we aim to investigate the effects of BMP-6 on OGD-induced apoptosis in human neural stem cells (hNSCs) and further explore the underlying mechanisms. The results may shed light on the functional role of BMP-6 in the ischemic brain injury and NSC therapy.
Materials and Methods
Clinical specimens
Tissues for hNSCs were dissected from the fetus cortex ranging in age from 12 to 15 gestational weeks and the detailed information was shown in the Supplementary Table S1. All the fetuses were obtained from the Affiliated Hospital of Xi’an Medical College with informed consent. The experimental protocols were approved by the Ethics Committee of Xi’an Medical University followed the guidelines of the Declaration of Helsinki. The specimen collection was conducted following the guidance of National Institutes of Health. No evidence of disease or abnormalities was observed after ultrasound and neuropathological examination of fetal brains.
hNSCs culture
Embryo brain tissues were collected in chilled sterile phosphate-buffered saline (PBS) and transported on ice. After removal of the meninges, the cortex was dissociated mechanically and incubated in dissection buffer (0.025% trypsin, 200 μM EDTA, and 2 mg/ml DNAse I in PBS, Sigma-Aldrich, USA) for 3 min at 37℃. Brain tissue was then mechanically dissociated into a single-cell suspension using a sterile Pasteur pipette and filtrated with a 40-μm cell strainer (BD Falcon, USA) followed by centrifugation at 1,000 RPM 4℃ for 3 min. Cells were collected and seeded at 150,000 cell/ml in non-adhesive T25 flasks (Corning, USA) with serum-free complete medium (DMEM/F12) supplemented with 1% N2, 2% B27, 20 ng/ml epidermal growth factor (EGF), 10 ng/ml basic fibroblast growth factor (bFGF) and 1% penicillin, 1% streptomycin (all from Invitrogen, USA) and incubated at 37℃ in a humidity incubator (Sanyo, Japan) with 5% CO2/95% air. Neurospheres were passaged to remove debris for the subsequent experi-ments. For single-cell adhesive culture, neurospheres were dissociated into single cells using TrypLE (Invitrogen) and plated in poly-D-lysine-coated 24-well or 6-well plates in complete medium overnight before use.
Oxygen-glucose deprivation treatment
Adherent hNSCs grown in a regular CO2 incubator. Two days later, the culture medium was removed, and cells were washed twice with pre-warmed (37℃) glucose-free DMEM medium (Life Technologies, USA). Deoxygenated glucose-free DMEM was equilibrated as the incubation medium, which has been bubbled with 5% CO2, 0.3% O2 and 94.7% N2 for 30 min prior to use. The cultures were then transferred to a hypoxia workstation (Bugbox; Ruskinn Technology, UK) and equilibrated with 5% CO2, 0.3% O2 and 94.7% N2 at 37℃. Hypoxia was induced by incubating cells at 37℃ for different time points (15, 30, 60, 120, or 180 mins) in the hypoxia workstation. After the OGD exposure, cells were removed from the workstation, washed twice with pre-warmed Hank’s balanced salt solution (HBSS, Life Technologies, USA), and incubated in above described hNSC culture medium at 37℃ in a humidified atmosphere containing 5% CO2/95% air for another 2 h. The control cultures were incubated at 37℃ for the same duration in a humidified atmosphere of 95% air/5% CO2.
Experiment treatment
To detect the effects of BMP-6 in hNSCs, cells were pretreated with a serial of concentrations of BMP-6 (0, 1, 10, 50, 100 and 200 ng/ml; R&D Systems, UK) for 6 h. Then cells were processed to OGD treatment, during which BMP-6 was added in the deoxygenated glucose-free culture medium in the BMP-6-treated group. Thirty minutes after the OGD exposure, cells were harvested for CCK-8 assay to assess the cell viability, and TUNEL staining to detect apoptosis. To abolish the effect of BMP-6 and p38 MAPK, noggin protein (3 μg/ml, the BMP‑6 inhibitor, Abcam, UK) and SB203580 (10 μM, the p38 MAPK-specific inhibitor) were added in the medium, respectively. For the negative control, cells were treated with the same volume of solvent only. All the experiments were performed in at least three independent hNSC cultures.
Cell viability assay
Cell viability was determined using the Cell Counting Kit-8 (CCK-8, Sigma-Aldrich) assay. In brief, adherent cultured hNSCs were grown in poly-L-lysine coated 96-well plates and following the experiment treatments, 20 μl of CCK-8 was added in each well to incubate for 2 h. The absorbance (OD value) was detected at 490 nm using a multi microplate spectrophotometer (BioTek, United States). Triplicate parallel wells were evaluated in all tests, and the data were obtained from the average of at least three independent experiments. The results are presented as the OD value.
TUNEL staining
Following the experiment treatments, apoptotic cells were detected using In Situ Cell Death Detection Kit POD (Roche, Germany) in accordance with the manufacturer’s instructions and cells were further counterstained with DAPI (1 mg/ml) before mounting. The positive stained cells were observed using a BX51 fluorescent microscope equipped with a DP70 digital camera (both from Olympus, Japan). Ten random fields on each sample were counted using a 20x objective. Data were presented as the percentage of TUNEL-positive cells in the total number of cells (DAPI-stained cells).
Immunostaining
The cultured NSCs were fixed in 4% paraformaldehyde solution (PFA) for 20 min, and 3 samples for each group were used for immunostaining. After three washes with PBS, the cell coverslips were permeabilized with 0.3% (v/v) Triton X-100 (T8787) for 30 min, rinsed, and blocked with 5% normal goat serum (Sigma-Aldrich) and 5% bovine serum albumin (BSA, Sigma-Aldrich) for 2 h. Then, the cells were incubated with the primary antibodies at 4℃ overnight. The following primary antibodies were used: mouse anti-nestin monoclonal antibody (1:200, Millipore), rabbit anti-SOX2 polyclonal antibody (1:200, Abcam, UK), rabbit anti-GFAP polyclonal antibody (1:200, Abcam), mouse anti-Tuj1 monoclonal antibody (1:200, Abcam), rabbit anti-HIF-1α polyclonal antibody (1:400, Abcam). All primary antibodies were diluted in 0.01 M PBS plus 2% BSA. For negative control, slides were incubated in PBS instead of primary antibodies. After washing, the cell coverslips were incubated with the following secondary antibodies: Alexa Fluor 488 donkey anti-mouse IgG (1:500; Invitrogen), Alexa Fluor 594 goat anti-rabbit IgG (1:500, Invitrogen). The nuclei were visualized by DAPI (H-1200, Vector, Burlingame, California, USA) staining. Immunostained positive cells were observed using a BX51 fluorescent microscope equipped with a DP70 digital camera (both from Olympus).
Western blotting analysis
Total protein was extracted from the cultured hNSCs using RIPA lysis buffer (Pierce, USA) complemented with a protease inhibitor cocktail (Roche, Germany), fractionated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA). Then, membranes were blocked for 1 h in 5% non-fat dry milk in Tris-HCl buffer containing 0.05% Tween-20 (TBST), followed by incubation with primary antibodies overnight at 4℃. The following primary antibodies were used: rabbit anti-procaspase-8 monoclonal antibody (1:1,000, Cell Signaling Technology), mouse anti-procaspase-9 monoclonal antibody (1:1,000, Cell Signaling Technology), rabbit anti-procaspase-3 polyclonal antibody (1:1,000, Cell Signaling Technology), rabbit anti-Cleaved caspase-3 polyclonal antibody (1:1,000, Cell Signaling Technology), rabbit anti-phospho-p38 polyclonal antibody (1:1,000, Cell Signaling), rabbit anti-p38 polyclonal antibody (1:1,000, Cell Signaling), and mouse anti-β-actin monoclonal antibody (1:10,000, Sigma-Aldrich). After washes with TBST, membranes were then incubated in horseradish peroxidase-conjugated anti-mouse or anti rabbit IgG (1:100,000, Sigma-Aldrich) for 2 h at room temperature. Immunoreactive bands were visualized using the enhanced chemiluminescent substrate (Pierce) and luminescent signal was detected by X-ray film (Fujifilm, Japan). The data were collected using a G:box gel imaging system (Syngene, UK) and analyzed using ImageJ software (NIH). The housekeeping β-actin was used as the internal control to normalize levels of target proteins.
Statistics
Statistical analyses were performed using GraphPad Prism software version 5.0 (GraphPad Software Inc, La Jolla, CA, USA). The data are presented as mean±standard deviation (SD) from at least 3 independent experiments. Significant differences between two groups were deter-mined using the Wilcoxon test for continuous variables while the Chi-squared test for categorical data. Differences among groups were analyzed using one-way analysis of variance (ANOVA), followed by Tukey’s post-hoc test. p<0.05 was considered statistically significant.
Results
OGD treatment induces apoptosis and hypoxia in cultured hNSCs
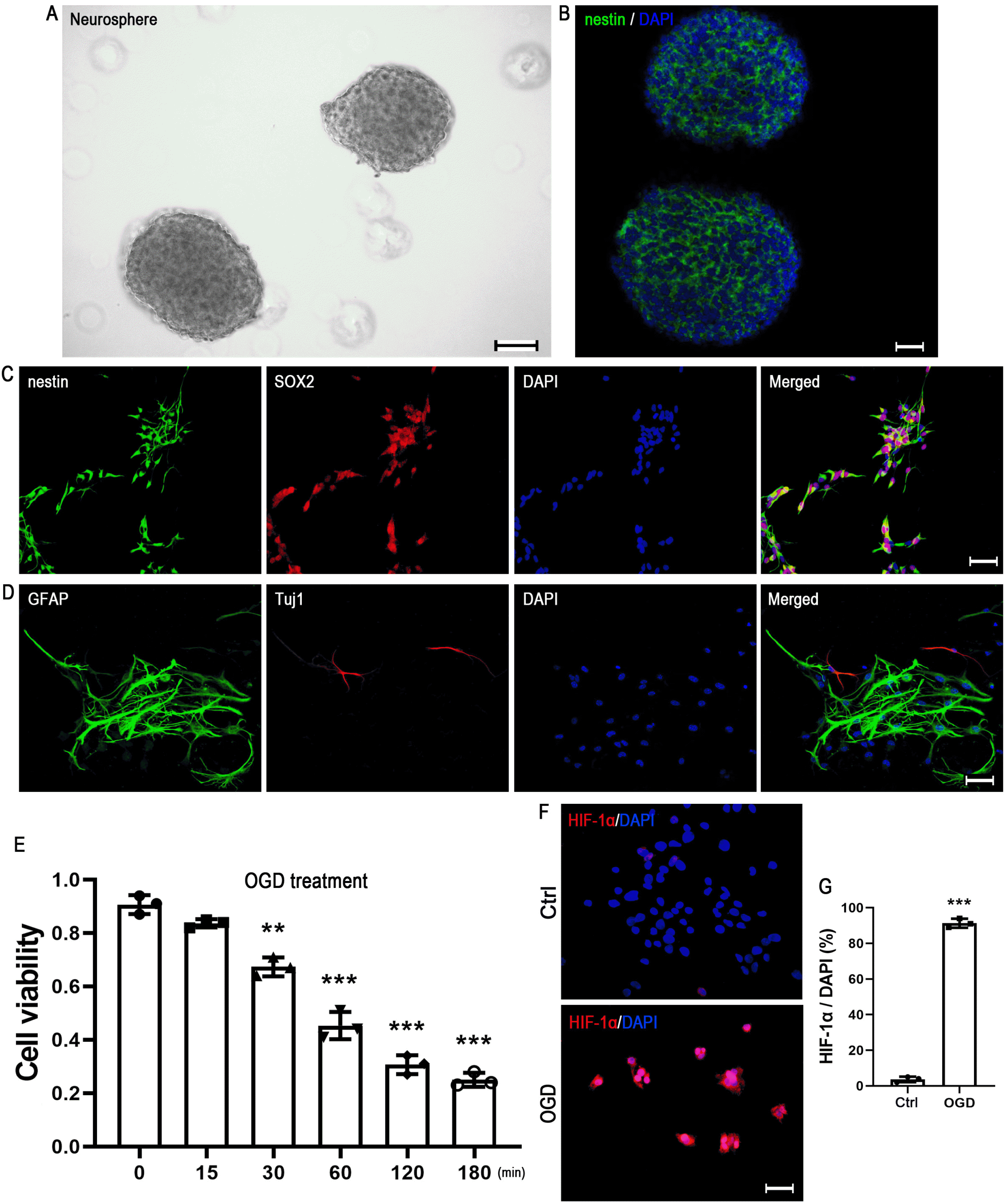
The primary hNSCs were dissected from four fetuses’ cortex which ranging in age from 12 to 15 gestational weeks and the detailed information was shown in the Supplementary Table S1. After 3∼5 days of culture, neurospheres with a diameter of about 100 μm could be seen in the medium (Fig. 1A) and most of the cells were nestin-positive (Fig. 1B). The NSCs specific marker nestin and SOX2 were used for further identifying hNSCs by immunofluorescence double staining. The results showed that 96% nestin-positive cells were co-stained with SOX2 (Fig. 1C). To identify the differentiation potential of hNSCs, single cells were cultured in the normal differentiation medium (DMEM/F12 supplemented with 1% N2, 2% B27 and 1% FBS, lacking bFGF and EGF). Five days later, astrocytes marker GFAP and neuron marker Tuj1 could be detected by immunostaining (Fig. 1D). In the present study, OGD treatment was applied as an in vitro model for hypoxic-ischemic injury. The CCK-8 assay showed that OGD treatment could significantly decrease cell viability in a time-dependent manner (Fig. 1E), and the remarkable damage effect was observed at 30 min or later. Since OGD exposure for 60 minutes resulted in a more obvious decline in cell viability, but did not lead to excessive cell death, we applied OGD treatment for 60 minutes in the subsequent experiments. To further confirm the OGD-induced hypoxia effect in hNSCs, HIF-1α immunostaining was performed to detect hypoxic cells. Compared to the control, the number of HIF-1α-positive cells was significantly increased after OGD treatment (Fig. 1F and 1G).
BMP-6 protects hNSCs against OGD-induced cell death
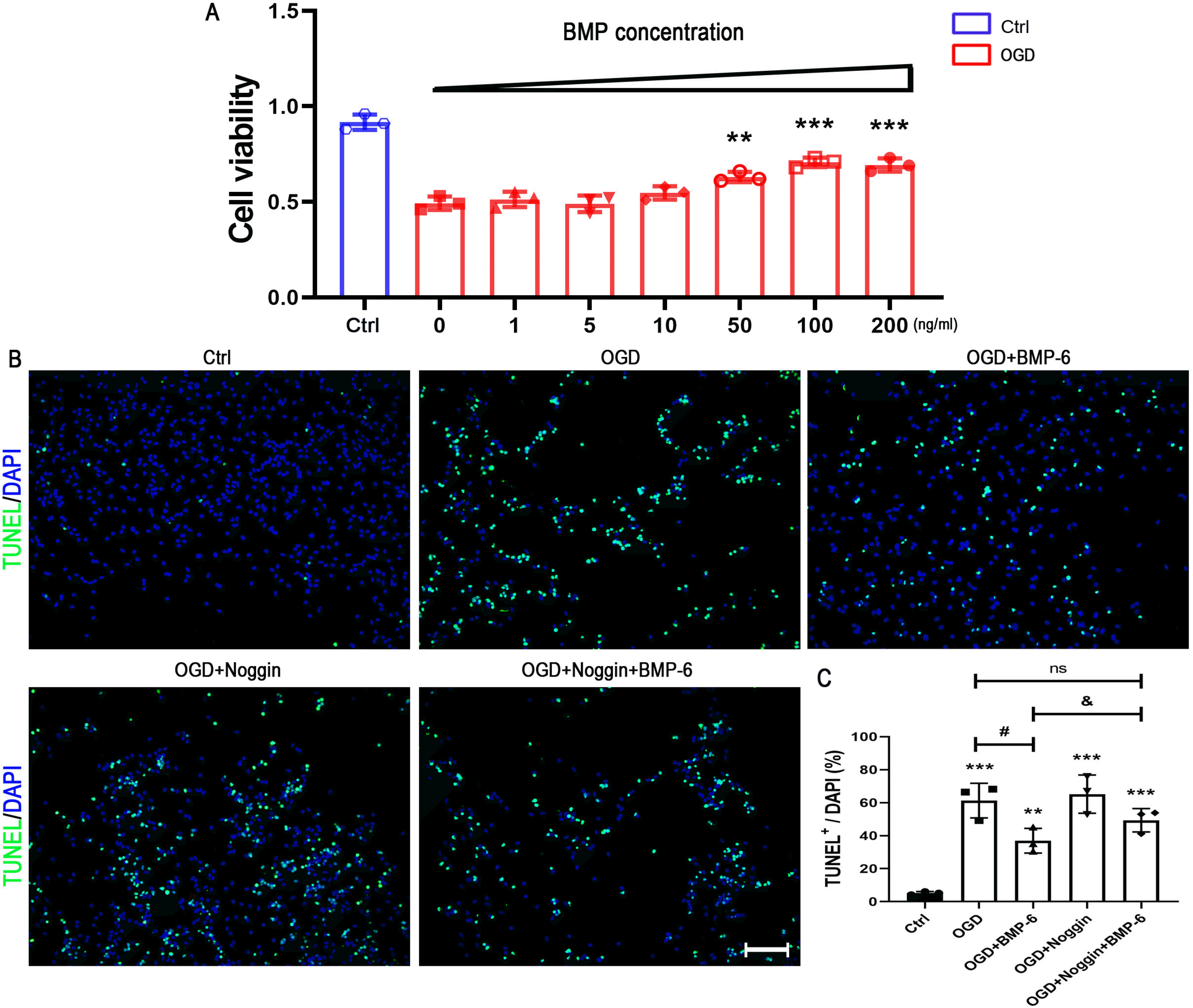
To determine the effects of BMP-6 in the OGD-induced hNSCs injury, cells were pretreated with different concentrations of BMP-6 (0, 1, 5, 10, 50, 100, or 200 ng/ml) followed by OGD treatment. CCK-8 results showed that BMP-6 treatment weakened the OGD injury in a dose-dependent manner (Fig. 2A). Because there was no significant difference between 100 and 200 ng/ml of BMP-6 treatment, 100 ng/ml of BMP-6 was used for the further experiments. TUNEL staining was applied to distinguish the apoptotic cells. It showed that the percentage of TUNEL-positive staining was about 60% after OGD treatment, while BMP‑6 significantly decreased the number of TUNEL-positive cells. Interestingly, noggin protein, the antagonist of BMPs, could attenuate the neuroprotective effects of BMP-6 on the OGD injury, as indicated by the higher positive rate of TUNEL staining (49.36%) in the OGD+Noggin+BMP-6 group than that (36.95%) in the OGD+BMP-6 group (Fig. 2B and 2C).
BMP-6 suppresses OGD-induced activation of apoptotic signaling
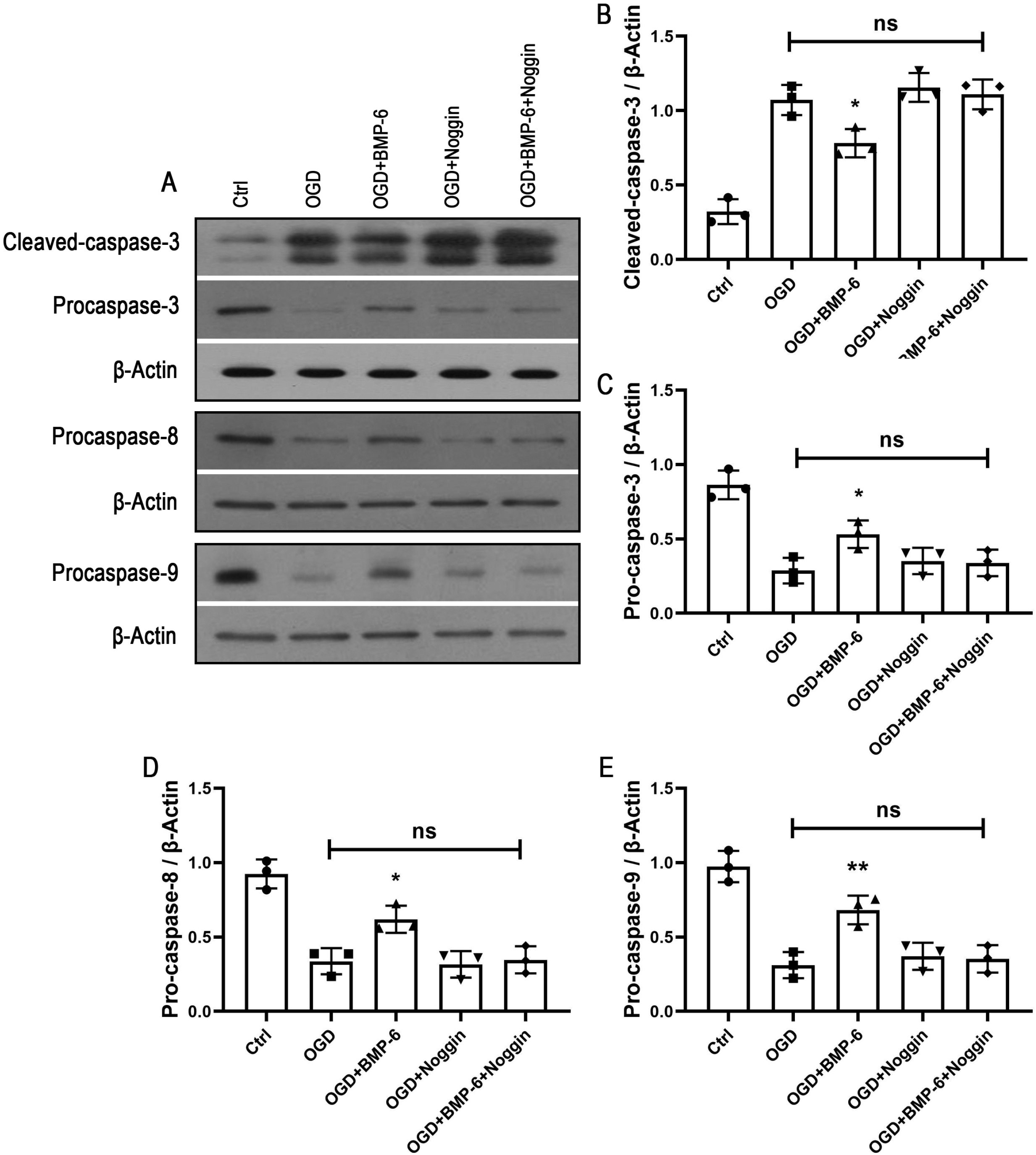
Several apoptotic proteins mediate cell death in HIBI, including proapoptotic precursor procaspase-8/9/3 (23, 24). To investigate whether the apoptosis-associated proteins are involved in the inhibitory effect of BMP-6 on the OGD-induced hNSCs apoptosis, the expression levels of cleaved-caspase-3 and proapoptotic precursor procaspase-8/9/3 were detected by Western blot analysis. The results indicated that OGD treatment markedly decreased the levels of procaspase-8/9/3 and increased the expression of cleaved-caspase-3, while BMP-6 effectively reversed the OGD-induced alterations in hNSCs. The effect of BMP-6 in the OGD-induced apoptosis was significantly attenuated by noggin protein (Fig. 3A∼E). The above data further revealed the anti-apoptotic role of BMP-6 in the OGD-treated hNSCs.
p38 MAPK signaling pathway is involved in neuroprotective effect of BMP-6 in hNSCs
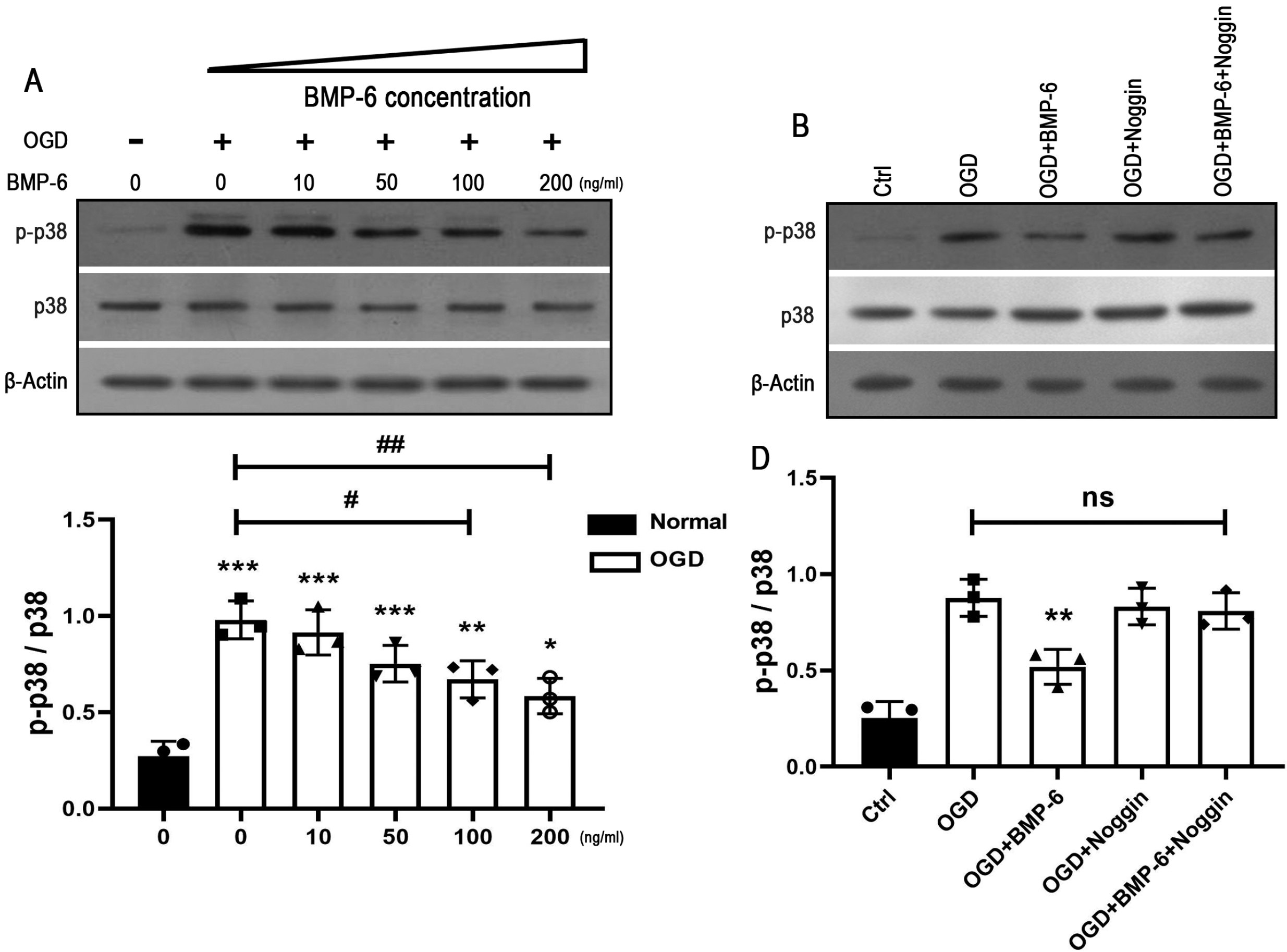
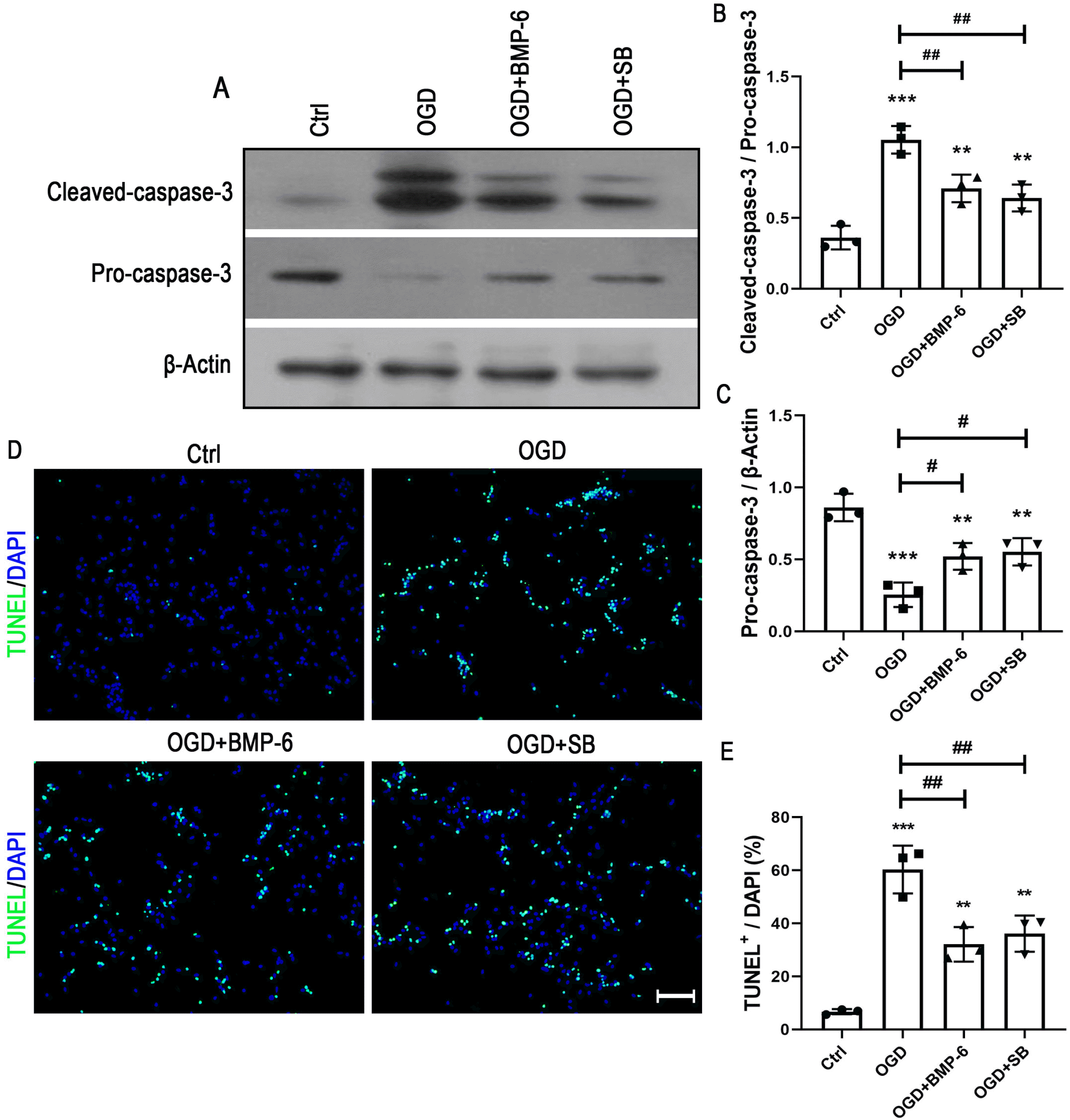
To investigate the intracellular pathway(s) in charge of neuroprotective effect of BMP-6 on the OGD-induced cell death, the activation of p38 MAPK signaling was detected by Western blot analysis. The results showed that OGD treatment markedly activated p38 MAPK pathway, while BMP-6 inhibited the phosphorylation of p38 in a concentration-dependent manner (Fig. 4A and 4C). Pretreatment of noggin efficiently abolished the effect of BMP-6 on the p38 activation (Fig. 4B and 4D). To further confirm whether p38 MAPK signaling pathway contributes to the neuroprotective effect of BMP-6 on the OGD-induced apoptosis, the additional experiments were performed in presence of the selective p38 inhibitor (SB239063, 10 μM). The caspase-3 assay showed that SB239063 could recover the OGD-induced alteration in the ratio of procaspase-3 to cleaved-caspase-3 (Fig. 5A∼C). Compared to the OGD group, SB239063 markedly decreased the number of TUNEL-positive cells (Fig. 5D and 5E). Besides, SB239063 showed similar tendency with BMP-6 in protecting hNSCs against the OGD-induced cell death. These results indicated that blocking the phosphorylation of p38 might contribute to the neuroprotective effect of BMP-6 against the OGD-induced injury in hNSCs.
Discussion
As a kind of multi-functional growth factor, BMPs are involved in many developmental processes including body axis determination, bone and cartilage formation, tissue morphogenesis, germ layer specification, and cell-fate specification (25, 26). BMPs can initiate signaling pathways by binding cooperatively to both Type I and Type II transmembrane serine/threonine kinase receptors (BMPRI and BMPRII) (27). With the in-depth study, there is mounting evidence that BMPs also play a role in preservating neuronal structure and/or function. Tsai et al. (28) found that BMP-7 had a neuroprotective effect in the culturd neuron, and BMP-7 overexpression not only protected neuron from oxidative stress but also attenuated lipopolysaccharide (LPS)-induced neuronal damage. In the transient ischemia and reperfusion model, administration of BMP-6 significantly reduced neurological deficit and infarct area (29). Moreover, BMP‑6 could protect retinal pigment epithelial (RPE) cells from H2O2-induced injury (21). In this study, we reported that BMP-6 markedly reduced the OGD-induced hNSCs apoptosis, as indicated by the decrease in the number of TUNEL-positive cells, increase in the procaspase-8/9/3 expression, and preservation in the ratio of procaspase-3 to cleaved-caspase-3. Moreover, the neuroprotective effect of BMP-6 might be mediated by p38 MAPK signaling pathway.
In the adult mammalian brain, NSCs are bounded in two specific brain niches: the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) in the dentate gyrus of the hippocampus. The special microenvironment of NSCs may be one of reasons for this phenomenon (30, 31). This niche consists of cells, extracellular factors, matrix glycoproteins, and blood vessels (32). Previous research revealed the increased release of inflammatory cytokines, overproduction of ROS, impairment on glutamate metabolism and cascades of interactive events in HIBI (33). And all of these factors could disturb the fragile balance in the niche, leading to the apoptosis of NSCs. Although transplanted or endogenous NSCs provide a novel treatment strategies for CNS disease, the hash niche limited its clinical application. Previous research (34) and our recent study have shown that BMP-6 can improve the capability of cells against hypoxia and ischemia insult, indicating an exploitable strategy for NSCs to withstand hash niche in HIBI. Therefore, BMP-6 might help to promote the clinical application of NSCs transplan-tation.
The recruitment of caspase-8 to the death-inducing signaling complex (DISC) leads to death receptor activation. Exposure of cells to reagents causing DNA damage promotes apoptosome formation and leads to caspase-9 activation (35). Both active caspase-8 and caspase-9 can induce the activation of downstream caspases, including caspase-3, and eventually cause apoptosis (36). Our study showed that BMP-6 pretreatment markedly suppressed the procaspase-3/8/9 activation and decreased the levels of cleaved-caspase-3, thus protecting hNSCs against the OGD-induced apoptosis. However, the link between BMP-6 and caspases activation remains unclear. Several studies suggested that BMP-6 could regulate the MEK/ERK/CREB/Bcl-2 pathway and induce the inhibition of caspase activation (20). In the present study, we found that inhibition of p38 using SB239063 exerted a similar effect with BMP-6, indicated as the decreased level of cleaved-caspase-3 in the OGD-induced hNSC injury. ERK and p38 signaling pathways interconnect closely at multiple levels (37, 38). Therefore, it indicated that BMP-6 might regulate caspases activation via p38 MAPK signaling pathway.
The intracellular signalling pathway help transmit signals from the cell surface to the nucleus, thus regulating gene expression in response to extracellular stimuli. As one of MAPK families, p38 signaling is preferentially activated by intracellular factors or extracellular environmental stresses, such as UV radiation, X‐rays, heat shock, inflammatory cytokines, and oxidative stress, and plays a key role in the regulation of proliferation, differentiation and apoptosis (39-41). Our present study showed that the phosphorylation of p38 was significantly increased after OGD treatment, and p38 inhibitor exerted a remarkable neuroprotective effect in the cultured hNSCs, which were similar with that of BMP-6. Moreover, BMP-6 could suppress the OGD-induced activation of p38. These phenmona suggested that the neuroprotective effect of BMP-6 might be mediated by blocking p38 activation. Nevertheless, p38 MAPK signaling pathway might interact with other signaling pathways including JNK (42), Akt (43), and NF-κB (44), as a complicated intracellular signal network. For instance, p38 MAPK can be activated by MKK3 or MKK6, and subsequently potentiates the downstream signals or activates other proteins directly (45). ROS oxidizes glutaredoxin to dissociate from ASK-1, resulting in the activation of JNK and p38 pathways (46). Intracellular signaling pathway is an intricate biochemical network, and different pathways interweave and influence each other, ultimately regulating cell behaviors. Whether other signal pathways mediate the above-mentioned neuroprotection remains to be investigated.
In conclusion, BMP-6 pretreatment protected hNSCs against the OGD-induced hNSC apoptosis, in which regulation of p38 MAPK signaling pathway may be one of the potential intracellular mechanisms. Our study displays a potential for BMP-6 as a drug target in treating HIBI and provides a novel therapeutic strategy for enhancing NSCs survival in OGD injury.
Supplementary Materials
Supplementary data including one table can be found with this article online at https://doi.org/10.15283/ijsc21093.
 XML Download
XML Download