

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) is an impressively adaptable analytical technology that provides significant benefit in clinical analysis [1]. LC-MS/MS has facilitated improvements in quality and throughput for numerous diagnostic assays, including steroid, amino acid, vitamin, peptide, protein, neurotransmitter, cancer biomarker, therapeutic drug monitoring, and toxicological assays [2]. There are numerous publications on novel approaches and assays deployed in the clinical environment, particularly focusing on development and the validation of outcomes. While these articles are of interest to clinicians and laboratory staff, there is a lack of publications on the operational lifespan of LC-MS/MS in the clinic.
Many laboratory processes are applicable to all technologies used in clinical analysis. Activities related to pre-analytical observations, documentation, or electronic medical records apply to the laboratory. Among guidance and best practice documents, resources are similarly broad, with some notable exceptions focused on LC-MS/MS, which are addressed further in this review. I will discuss components of validation and operation specific to LC-MS/MS, with a focus on those aspects of the technology that are true differentiators in clinical analysis.
This review is a follow-up of a previous one [3]. Together, these publications intend to capture the lifecycle of LC-MS/MS from development through validation to the execution and maintenance of a clinical assay. Despite best efforts, no mechanism exists to encapsulate the specifics of individual assays, laboratory setups, or procedures. However, the principles discussed herein are broadly applicable to the processes, experiments, and protocols used to generate high-quality data from clinical LC-MS/MS assays. Readers should note that sections are commonly cross-referenced within this review, reflecting the nature of LC-MS/MS workflows in that practices occur in parallel. For example, data review during calibration verification is a nested process; each subsection represents part of a whole of clinical LC-MS/MS analysis and should be read as such.
VALIDATION
Validation has several colloquial meanings, but the International Organization for Standardization (ISO) defines validation as “confirmation, through the provision of objective evidence, that the requirements for a specific intended use or application have been fulfilled” [4]. Regardless of the technology, the validation of quantitative lab tests must demonstrate accuracy, precision, stability, analytical specificity, linearity, and lack of interferences [5]. To support the claims of intended use of LC-MS/MS assay results, the list of validation experiments can be long. Literature reports vary widely in how validation experiments are to be executed. Demonstration of all experimental outcomes of validation may not be necessary to support the hypothesis in a publication. Alternatively, word count limits may constrain exhaustive descriptions of validation assays in reports. Consequently, critical experiments for LC-MS/MS validation may not be fully disclosed in the numerous reports.
Resources for validation guidance are issued by multiple institutions. The US Food and Drug Administration (FDA) Bioanalytical Method Validation (BMV) guidelines prescribe drug development assays focused on MS analysis [6]. In the most recent version of the BMV, specific expectations and guidance on how to address certain endogenous biomarker concerns, such as the use of stripped matrices and commutability, are further clarified [7]. However, the FDA BMV is not designed for diagnostic laboratories and has discrete principles and practices (e.g., incurred sample re-analysis experiments) suited for different purposes. Nevertheless, many experiments utilized for the validation of clinical diagnostic assays are mirrored in these guidelines.
An entire chapter of the Tietz Textbook of Clinical Chemistry is dedicated to a distillation of the FDA BMV, CLSI guidelines, and general best practices for the execution of LC-MS/MS validation [8]. It prescribes batch sizes, replicates, designs, and timeframes for validation experiments. Furthermore, a review of experiments and acceptance criteria has been recently published [9]. As experimental designs, timeframes, and degrees of replication have been addressed elsewhere, this review focuses on the more enigmatic components of LC-MS/MS validation for diagnostic testing.
Dilutional integrity in validation
The establishment of dilutional integrity is fundamental to quantitative assay validation. Approaches differ in the predilution concentrations used for assessment. In one approach, samples are fortified to a concentration above the analytical measurement range (AMR) and measured only after dilution to within the AMR [10, 11]. Alternatively, samples measured to lie within the AMR are diluted and re-assayed [12, 13]. Both approaches are accepted in practice and have their limitations. In the case of fortification above the AMR, errors in sample preparation, changes in analyte equilibrium, and chemical modifications can lead to false conclusions. For example, in free homocysteine measurement, the fortification of free homocysteine in human plasma results in under-recovery as the thiol binds to other free thiols in the sample [14]. Pre-analytical sample manipulation is required to achieve accurate analytical homocysteine measurements when samples are diluted in validation experiments. These conditions may not represent the true matrix intended for use in the assay. In the absence of this knowledge a priori, a failure of dilutional recovery of homocysteine may be ascribed to an analytical flaw. Alternatively, accidental over-spiking may result in a false-positive observation of dilutional linearity without a means to detect the error.
Dilutional integrity assessment using a sample within the AMR can avoid such issues as it is less reliant on accurate recovery. The sample to be diluted is measured in the same batch as the diluted samples. Data reduction is then relative to the measured concentrations of the diluted and undiluted samples, without a reliance on the trueness of fortification. This approach may not mimic the exact conditions of excess analyte. Errors associated with supra-AMR concentrations, such as an overload of extraction medium or ionization competition with an internal standard (IS), may not be identified [15, 16]. However, these deviations are readily observable during data review (see the “Post-acquisition data review” section below) and result in sample re-assessment upon further dilution. In such cases, absolute recovery of a supra-AMR sample should not be assumed; however, few clinical assays require absolute determination of concentrations significantly higher than the upper limit of the AMR. If this is required, the method should be re-developed for calibration at the clinically meaningful levels.
Imprecision design in validation
Recent CLSI guideline updates suggest that objective evidence has to be provided for laboratory-developed tests just like it is expected from in-vitro diagnostic device providers [17]. In guideline documents, LC-MS/MS assay method developers are regarded “manufacturers.” This may result in the reframing of previous expectations of a laboratory’s desirable goals and experiments. For example, 20 days of experimentation for precision are preferred over a three- or five-day approach as 20 days of evaluation are more likely to allow the adequate definition of measurement uncertainty or identification of failure points in the protocol [18]. Given that isotopically labeled ISs and batch-based calibration can be used for normalization, sources of variance from the operator(s), calibration cycle, instrument, or reagent lot should be readily identifiable during critical review of the validation data (see section “Post-acquisition data review” for details). The commonly utilized three-day evaluations for precision and accuracy in LC-MS/MS validation were derived from bioanalytical clinical trials, some of which are utilized for only a few months of analysis [6]. In contrast, a validated LC-MS/MS assay for diagnostic testing can be used for many years.
Limit of blank (LOB) and limit of detection (LOD) in validation
Some guideline-recommended experiments are difficult to rationalize when applied to LC-MS/MS validations. The experimental design and the usefulness of the data must be viewed in the context of the technology. For LC-MS/MS, a consideration is the use of response (signal) versus a concentration (most often derived from an analyte-to-IS ratio and a calibration curve). The former is always affected by instrument performance and is largely irreproducible between batches while the latter can be more rugged against instrument performance, especially at higher signal. The distinction in data is significant as LC-MS/MS performance changes over time [19]. Ion lens contamination, source fouling, mobile phase solvent/additive quality, a change in vendor for a critical reagent, random occurrences of phthalates, and many other issues can contribute to the loss of signal or an increase in noise between samples, batches, runs, or days [20-22]. Additionally, concentrations below the lower limit of quantitation (LLOQ), as implied in the term, have undefined imprecision, rendering absolute determination unreliable.
As described in CLSI EP17, the LOB and LOD are difficult to determine [23]. The raw signal associated with the quantity of a compound is highly dependent on the fitness of the mass spectrometer at the moment of analysis. A recently cleaned mass spectrometer will yield a different LOB/LOD than one that has been extensively used. Assays commonly relate the LOD to the LLOQ and entirely ignore the LOB, particularly when the LLOQ is included in each batch sequence [24-26]. The recommendation to include an LLOQ sample in each batch is sufficient evidence for the recognition of this actuality [6].
The determination of the LOB and LOD may be confounded by endogenous analyte concentrations or common LC-MS/MS calibration schemes. Preferably, the matrix in LOB/LOD determination experiments is equivalent to the intended sample type to account for matrix effects. If an endogenous amount must be diluted to achieve concentrations lower than the lowest calibrator, the modified sample may have significantly different matrix effects. Similarly, LOB/LOD determination in a solvent-based calibration system assumes that the ionization cross-section is equivalent between the solvent and real matrix. For example, supplemental data for a recent testosterone assay demonstrated a difference in ionization suppression between stripped serum and patient serum [27]. These two sample types would have very different LOB/LOD values as the response function of true serum was lower than that of the stripped serum used in the validation experiment. It should be recognized that LOB/LOD assessment of authentic patient samples may be utterly important if such claims are desired. However, given the performance variation, assuming the LOD to be equivalent to the LLOQ and the LOB to be undefinable are logical conclusions for LC-MS/MS assays.
The signal-to-noise (S/N) ratio is another confusing metric when applied to LC-MS/MS validation. Historically, MS-related S/N ratios were associated with peak identification/detection rather than quantitation based on peak areas. Consider an assay developed for a 5-µm particle being used with a 1.7-µm particle, without any other modification to the assay. For equivalent injections, the S/N ratio would be improved because of improved chromatographic efficiency, but the absolute area should not change. Thus, any quantitative difference would only exist as a function of imprecision in integration (differentiating peak from noise for the lower bound of the integrated area). There is no intrinsic link between an assay’s LLOQ as a function of quantitative precision and accuracy and the S/N ratio [28, 29]. Additionally, noise in an MS/MS chromatogram can be due to various variables than other instrumentation. The sequential scanning aspect of data acquisition and the averaging of signal (counts/sec) can complicate noise determination, especially when using scheduled MS/MS [30]. Finally, data reduction software often use smoothing algorithms to achieve more reliable peak integration. Bioanalytical guidelines do not indicate whether the S/N ratio is best calculated prior to or after smoothing or what degree of smoothing is allowable.
The previous two sections on the LOB/LOD and S/N ratio raise the question “What claim of the assay is objectively verified?” If a clinically meaningful value is persistently detected at a quantity below the LLOQ, the laboratory staff may find it difficult to report unambiguous results. If this occurs frequently, it is appropriate to return the assay to method development for the establishment of a calibration range sufficient for measuring the relevant concentration range.
Acceptance criteria in validation
Acceptance criteria for validation experiments can be obtained from various sources. For diagnostic assays, broad guidelines have been issued by the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) [31]. These guidelines describe “how good” an assay needs to be. Three general methods are used in the EFLM model. In the most preferred method, the analytical performance is defined based on the determination of its effect on clinical outcomes. This can be indicated by well-controlled experiments or by consensus of expert opinion. Second, total error limits can be applied as a function of biological variation (BV). In the third approach, criteria are based on the state of the art, which is defined as “the best achievable with the current technology.”
Most LC-MS/MS assays fall within the third group (performance criteria based on the state of the art) and apply imprecision and inaccuracy targets of <15% at non-LLOQ concentrations and ±20% at the LLOQ. This is derived from historical values associated with LC-MS/MS in clinical trials according to the FDA BMV and the European Medicines Agency guidelines [6, 32]. These targets may be reduced when BV is generated and applied to the measurand. For example, for testosterone harmonization, which is part of the Hormone Standardization Program (HoSt) established by the US Centers for Disease Control, the criterion for harmonization was determined to be a mean bias of ≤6.4% when measured in 40 samples across the laboratory’s measurement range [33]. Therefore, validation specifications for testosterone must maintain a mean bias <6.4% for confident harmonization. These criteria allow for instances in which a laboratory could be imprecisely accurate by virtue of the average and maintain accreditation in the program only because of luck and/or randomness. The reader should recognize that neither luck nor randomness are preferred attributes of clinical assays.
As for BV, the values for inter- and intra-individual variation (CVi and CVg, respectively) may have interesting origins. Based on an understanding of the source of total allowable error, BV expectations may be modified in the context of the assay’s target population. Testosterone serves as an excellent example. The CVi and CVg were determined based on a compilation of four studies [33]. These studies utilized a small, homogeneous population of only 43 individuals to estimate the BV in the global population. Specifically, all study participants were male, described as “healthy,” and based on their country of origin (Spain, Belgium, Finland, Denmark), of European descent and most likely, Caucasian [34-37]. In addition, of interest is that immunoassays were used in each study. It can be assumed that these studies were executed by very capable laboratories using techniques that surpass standard laboratory assays; however, it cannot be definitively concluded that the challenges associated with steroid measurements in direct immunoassays were completely absent in the procedures used [38-40]. In short, the BV used to generate the testosterone HoSt criteria and ascribed to all possible patients was derived from the analysis of 43 adult males of European descent measured using assay technology that does not represent the current gold standard for this analyte. Therefore, the usefulness of the BV-derived criteria set forth in the HoSt program could be debated [41]. Are these criteria meaningful to the laboratory’s target population? Does the application of the BV-derived criteria increase the quality of the laboratory? Maintaining a 6.4% maximum allowable mean bias for testosterone requires exceptional control of calibration and test articles, beyond the implications of the actual procedure. However, such criteria are better than none and perhaps better than a generic 15% criterion.
POST-VALIDATION AND PREPARATION FOR LAUNCH
With a validated method in hand, the laboratory is not yet prepared to test patient samples. Prior to the launch of a new assay, assay translation must be addressed, particularly if the research/development/validation technicians are not actively working in routine patient testing. In a sense, the research/development/validation technicians can be treated as a distinct entity from the operations team, representing two different laboratories. Protocols for inter-laboratory method transfers have been outlined in the context of clinical trials or pharmaceutical analysis and provide important recommendations [42, 43]. These experimental components can also be utilized to satisfy regulatory requirements, such as competency evaluations for staff performing the tests, if the laboratory is subject to such obligations [4, 44].
In method transfer/competency, aspects beyond the extraction/analysis protocol should be assessed. All components of the procedure prepared in the testing facility benefit from this exercise, even if the performing staff are highly skilled and competent. This is because of the concepts of oral transmission of laboratory skills or expectations for integrated knowledge, which may not be translated in a standard operating procedure (SOP). The Aims and Scope of the Current Protocols series from Wiley present this point quite well: “nuances that are critical for an experiment’s success are not captured in the primary literature but exist only as part of a lab’s oral tradition” [45]. This may hold true for SOPs for assays that have not been operationally stressed or attempted by multiple technicians, who each have their own interpretation of written and unwritten laboratory processes.
The SOP must be revised during the transfer process to ensure that sufficient details are included, even if that knowledge is considered extant in the laboratory. Quality system expectations have recently included specific sections addressing knowledge management to ensure that critical information is retained both on paper and within the collective knowledge of the staff [46, 47]. An exercise may be performed wherein staff are asked to perform the assay as described explicitly by the SOP; each question asked by the staff during this process may represent SOP language worth expansion or clarification.
The competency assessment and transfer process should be structured to include critical reagent changes, new calibration material correlation, new quality control (QC) range generation, and patient sample correlation. This effort is similar to lot-to-lot verification, which is discussed in detail below. Broadly, scientists/technicians, calibrators, QCs, and instrumentation involved in validation would be used to compare the laboratory’s normal manufacturing and operational components. Patient samples are to be assayed in parallel with validation materials and samples freshly prepared in the operational environment (within the stability timeframe of the validation materials). Acceptance criteria include standard assay evaluations for batch acceptance, including clean blank samples and transition ratios within tolerance. Comparison of the results for the validation materials and operational samples can be expressed as imprecision/inaccuracy on a per-sample or all-group basis or be based on regression of the dataset (preferably, Deming or Passing–Bablok regression) and review of the slope and correlation [48]. Expectations for these comparisons should be in line with those defined in the assay validation, associated with state-of-the-art criteria or the BV of the measurand.
These data can also be utilized for the validation of laboratory information system (LIS) integration of a new workflow. Analyte identifiers, reference intervals, and reporting units (including decimal points and rounding) are all LIS features to be evaluated for new assay launches. Assay-specific determinations in the LIS, such as pertinent clinical history, scheduling of dilutions, modifications/calculations associated with the test result (e.g., creatinine normalization), and alert values, must be verified during the pre-launch phase [49]. These data should be reviewed and confirmed in a test environment prior to assay deployment.
OPERATION OF LC-MS/MS ASSAYS
General laboratory concerns, such as facility maintenance and the inspection of patient samples for pre-analytical deviation, are consistent for all laboratory assays and are therefore not discussed here, apart from mentioning that maintaining a significant quantity of reagents (within the expiry dates) is particularly essential for LC-MS/MS assays. It is not uncommon to have back-orders or material shortages of the many supplies required for testing, such as the global reduction of acetonitrile experienced between 2008 and 2011 [50, 51]. A sufficient supply allows the time required to source, evaluate, and validate necessary modifications to the method when a shortage becomes imminent. Thorough assay validations can indicate the viability of alternative critical reagents where appropriate [52].
Validation should also identify acceptable parameters for the assay to be run at scale within the laboratory. These include the maximum batch size (numbers of samples included in a single run), the frequency of calibration curves, and the number of samples between QCs. In clinical laboratories, it is common to prepare patient samples for LC-MS/MS in parallel, utilizing 96-well plates, multichannel pipettes, and multi-head liquid handlers [53]. However, validation may have been performed using different volumes from those used in the operational assay; in such cases, the laboratory has to determine the standard/QC frequency after the assay has been launched. Often, 96-well plate assays include calibration curve standards at the beginning of each plate and either reanalyze the standards or prepare another set of calibrators at the end of each plate (batch-calibration mode) [54-56]. Less frequent calibration is demonstrated for certain assays [57]. Careful evaluation of the instrument response drift and the implementation of re-calibration/maintenance when QCs fail are essential to realize the gains from non-batch-based calibration. The placement of calibration curve standards at defined intervals (e.g., batches, numbers of samples) allows for the prompt identification and remediation of issues with the assay. Additionally, consistency in calibration placement can reduce the burden on technical staff by providing a reliable expectation of the standard curve frequency. This allows for simple decision-making flow charts when a calibration curve fails to meet certain criteria.
QCs are fundamental to laboratory testing and applied to all types of assays. LC-MS/MS poses specific challenges in terms of QC. Some historical context is appropriate. The widely adopted approach of applying standard deviations to the results to monitor assay performance was originally discussed in 1950 [58]. In 1952, the concept was expanded with the adoption of a patient pool as QC material [59]. The treatment of QC results as used in current practice was published in 1977, 8 years before the first graphing calculator became commercially available [60, 61]. This means that all calculations (means, standard deviations) were likely performed manually using standard statistical tables. The multi-rule approach as described by the CLSI (i.e., 13S, 22S, R4S, 4IS, 10X determinations) is deployed in numerous clinical tests [62]. These QC treatments are representative of single-measurand assessments, whereas LC-MS/MS can measure multiple compounds in a single sample.
Multiplexed measurement is not uncommon in LC-MS/MS assays for diagnostic purposes. Numerous molecules can be measured in a single sample, regardless of their biological or clinical co-relevance. Amino acids and acyl carnitines by flow injection MS/MS, steroid panels, drug evaluations, neurotransmitter assays, and assays of classes of proteins are all multiplexed LC-MS/MS assays in clinical use [63-67]. If one concedes that statistical rates of randomness used in QC evaluations apply to LC-MS/MS assays, any analysis that measures more than 6–9 molecules presents great difficulties [68]. Multi analyte assays face challenges of compounding randomness (imprecision) that single-analyte assays do not [69, 70]. To address this, there are some practical approaches that can be considered when measuring multiple components from a single QC. These approaches consider the analytical control afforded by isotopically labeled ISs and the richness of LC-MS/MS data applicable to root-cause analysis of errors.
First, we will explore the features of QCs as a means of accepting a batch. A pass/fail status can be applied as a static bias from the expected concentration (measured or theoretical), which is typically 15% for non-LLOQ concentrations and 20% at or near the LLOQ, rather than being assessed from standard deviations from the mean [6, 32]. The establishment of expected concentrations is discussed in “Assay Maintenance” section. Precision is poorly accounted for as each QC analyte is assessed for inaccuracy in an acute manner. Longitudinal evaluations serve to assess drift in concentrations and, if warranted, shift the expected mean. However, consistent drift observed across numerous calibration events for all QCs may indicate a change in calibration or QC concentrations. Such drift must be investigated prior to modifying the expected QC concentration(s). If a QC is determined to fail due to gross error (e.g., undefined concentration because of no IS response), the data should not be included in the longitudinal dataset; all other data should be included. Broader or tighter criteria can be applied based on total allowable error for the molecule(s) of interest, although this would remain an acute determination for each batch [71]. Again, consideration of the number of co-measured molecules is necessary and may result in expansion of the acceptance criteria.
One approach to multiple-analyte QC has been to move state-of-the-art acceptance conditions from a 15% maximum inaccuracy to a 30% maximum inaccuracy [72]. A 30% error in urinary drug levels is typically of little consequence as the intended use is the detection of compounds; however, for assays where quantitation is clinically meaningful, such allowable inaccuracy may be inappropriate. Discussion with appropriate clinical groups responsible for interpreting the results of multiplexed analyses can provide insights into the required degree of reproducibility.
QC results may also be interpreted in the context of the results of other samples in the batch. For example, in a multi-analyte drug assay, a high-concentration QC fails for one drug of interest by measuring more than 115% of the expected value, but just outside of the acceptance range. Two lower-concentration QCs pass for the same analyte. All IS responses for the single analyte are within 20% of the mean of the calibrators for each sample. No sample in the batch produces a signal for the analyte of interest. In this case, there not necessarily is batch failure, and re-analysis is not required given that the low recovery is acceptable and the high-concentration QC failure does not indicate an error that would lead to false-negative results. The context of pass/fail assessments based on QC results in LC-MS/MS should be assay-dependent and leverage the capabilities of the platform for accurate reporting [73]. Given the analytical detail provided in LC-MS/MS, QCs can serve many purposes. The underlying role of QC materials in evaluating drift and error in an assay is key to any approach and must be considered as the minimal framework for QC result interpretation [62].
The frequency of QC analysis for LC-MS/MS analysis depends on numerous factors. Under the Clinical Laboratory Improvement Act (CLIA) issued in the US, a minimum of two liquid levels of QCs must be assayed every 24 hours of analysis for any technology [74]. However, such a QC scheme may underserve LC-MS/MS assays in error detection. Variation in absolute recovery can occur between batches of samples and response function drift is a recognized LC-MS/MS feature. While validation exercises can guide QC limits due to assay calibration drift (distinct from instrument mass calibration drift), other aspects of the laboratory work flow must be considered for risk [75]. For example, solid phase extraction (SPE) can use various versions of manifolds for performing both tube-based and plate-based SPE. The manifolds can hold 8, 12, 16, 24, or even 48 tubes; plate manifolds are often constructed for 96 or 384 wells. As a QC check against variable recovery due to a difference in manifold settings (e.g., excessive vacuum applied at sample loading), at least one QC/manifold/run is recommended in a tube-based SPE protocol. Plate-based SPE (96 sample capacity) would benefit from multiple levels within a single plate [76]. Similarly, batches of manual liquid-liquid extraction analyses would include QCs based on the number of tubes that can be assayed in a contiguous manner as determined by the space available in an evaporation device.
LC-MS/MS QUALITY ASSURANCE
Given the breadth and variety of meanings, a characterization of the term “quality assurance” (QA) is appropriate prior to a more detailed discussion. The International Union of Pure and Applied Chemistry (IUPAC) definition is perhaps most closely tied to our intention, in that QA is designed to protect against the failures of QC [77]. QA should also provide mechanisms to ensure the veracity of not just QCs, but all samples assayed. This encompasses blanks, standards, QCs and, most importantly, patient samples.
QA can be partitioned into evaluations of meaningful pre-analytical, analytical, and post-analytical metrics. These are included in the quality management section of the College of American Pathologist’s all Laboratory Common Checklist [78]. Each evaluation relies on distinct elements to determine whether a system is executing the measurement as intended. All elements should be considered in the context of successful and reliable sample analysis, with identified failures initiating investigation and remediation.
Pre-analytical QA
Aspects of pre-analytical quality management in terms of sample fidelity and capture of meaningful demographics have been discussed [79-82]. These components are appropriate for all laboratory test QA. For LC-MS/MS assays, pre-analytical QA takes place prior to sample preparation and can encompass a breadth of assessments, as shown in Table 1. It includes logical checks, such as “Are the mobile phase bottles full?” or “Is the correct column installed?” or “Have the calibrators expired?” Such examinations benefit from a checklist associated with a batch’s documentation, which is helpful during troubleshooting and root-cause analysis of errors. Specific to LC-MS/MS QA is the assessment of the analytical system prior to the extraction of patient samples. There is minimal value in preparing samples that cannot be analyzed due to problems with the LC-MS/MS system that require repair. Delays in the ordering of components or the time required in complex maintenance (e.g., rail-pull to resolve ion lens/quadrupole charging and subsequent return to vacuum and retuning) can sometimes take days.
As stated previously, the performance of LC-MS/MS systems changes over time. Columns can become fouled with injected material, providing operationally unsafe back-pressures. Stationary phase interactions are modified by use, yielding poor retention or less-than-desirable peak shapes. Ionization and ion transfer efficiency can be decreased due to contamination of the source and optics, resulting in lower response functions. These are all natural occurrences in the lifecycle of LC-MS/MS, and no working LC-MS/MS system is immune to them. It has been jokingly stated that “I’ve only seen one mass spectrometer act in a consistent manner between days and even weeks. And that mass spec was unplugged” [83].
This system check has been described in various forms and is generally known as the system suitability test (SST). Analyses utilizing chromatography and/or MS have such checks instituted as part of best practice or regulatory guidelines [6]. In some approaches, a generic set of compounds is utilized. These SST solutions contain analytes that are distinct from the measurand(s) of the assay and are measured to determine system performance [84-86]. In contrast, recommendations for targeted quantitative system suitability testing specify that the measurand(s) must be present in the solution [87]. This affords a more direct observation of the status of the system for its intended use.
SSTs provide an opportunity to devise action limits for data metrics [88]. These limits serve as triggers to perform some maintenance function, such as changing of the column due to degradation [89]. The depth of SST data is such that many maintenance aspects can be converted from unplanned maintenance after an error to a planned maintenance event. Preventive maintenance can reduce the need for re-injections and lower the downtime of a system. Predictive analytics in clinical testing are currently applied to a patient population or test results, providing a means to translate those processes to laboratory equipment [90]. With retrospective data interrogation from the validation experiment or operationalized tests, instrument-diagnostic cutoffs can be applied to analytical procedures for error prediction [91].
SST observations that result from errors may present in an immediate form, a longitudinal manner, or both. Take for example increased pressure across the LC system [92]. Increased back-pressure over numerous sample injections is normal. It is also common to observe an immediate loss of pressure (e.g., due to cracks in the tubing, leading to leakage of the injected samples/mobile phase) or an immediate increase in pressure (e.g., due to a blockade in the tubing or column). A malfunctioning signaling board/detector may be indicated by a slow decrease in the response function (requiring a change to a detector setting to correct) or a complete failure of the detector, each of which is easily detected by tracking of the SST data.
Each incidence of maintenance in LC-MS/MS has a data-derived “trigger,” whether or not those data originate from SST injections. For example, high back-pressure can be isolated to the LC column, which can then be reversed (and either flushed and returned to service or used in the reverse direction for analysis) or be disposed of [93]. Table 2 lists metrics that can be evaluated and the most common offending component(s). Note that both IS and analyte transitions can be informative of system performance, especially when they disagree in chromatographic metrics. In certain aspects, a visual review suffices to determine whether or not a system is operating within expectations. The absence of signal where it has previously existed should alert technicians that an error has occurred. A dramatic peak shape alteration, yielding large peak width values or significant variation from an asymmetry of 1 (i.e., <0.2 or >5), can be easily detected on screen. These acute determinations should initiate a troubleshooting investigation and correction.
In addition to immediate determinations of instrument performance, weekly/monthly monitoring of SST data offers enormous value for LC and MS upkeep [94]. Features of review are identical to acute pass/fail decisions for a batch of samples (e.g., LC back-pressure, retention time, peak areas) but should be interpreted by retrospective comparison. A trend line of data from previous SSTs can indicate a degradation of the investigated metric. A subtle change in day-to-day retention times may not be noticed on visual review, but a plot of such data may show a clear negative regression slope. A cut-off value could be applied to direct operators to install and test a new column. Similarly, defining a minimum performance expectation of analyte response can indicate to operators that the instrument has to be cleaned prior to further sample analysis. Data interrogation tools designed for non-targeted approaches may be adopted for assays with specific measurands [84, 95]. These tools can simplify the review of SST data to remove subjectivity in decision making.
One metric used in SSTs is the peak area ratio or raw peak area [96]. When the SST analyte is injected into the instrument at a meaningful concentration (i.e., the LLOQ) and measured in replicates (3–4), reproducibility can be determined. Importantly, measurement of the SST analyte in replicates can indicate imprecision of the area ratio, which all quantitation is based upon. Note that S/N ratios are not captured for reasons discussed in the validation section above. Significant changes in the imprecision of area ratios in an SST can indicate poor response function of the analyte at the LLOQ or possibly, a more nuanced instrument issue.
Following any instrument maintenance, SSTs can be used to requalify the platform. This also applies to maintenance as performed by external service engineers. Each MS manufacturer has minimum specifications for platform operation that can be achieved by an engineer subsequent to a repair or planned maintenance. However, vendor specifications are not suitable evidence for assay reliability. Assay-specific SSTs afford an objective benchmark for the intended purpose of the platform and should be assessed before the service engineer leaves the facility.
Intra-analytical QA
QA continues through the analytical phase of sample analysis. Four features have identified as primary causes of error during this phase, accounting for 7%–13% of all laboratory related errors [97]. These are equipment malfunction, sample mix-ups/interference, procedure not followed, and undetected failure in QC, although the actual proportions of these individual sources are undefined. Each of these errors apply to the LC-MS/MS workflow. Equipment viability is generally obvious in a workflow (e.g., a broken pipette or a non-operational evaporator have identifiable symptoms) and SSTs qualify system performance. Data review should easily identify a QC failure (bias/imprecision) or interference (transition ratio, chromatographic features). Of importance to LC-MS/MS are sample mix-ups and deviations in the procedure. Table 3 lists details to be captured to mitigate or reduce the associated errors.
Sample mix-ups can occur at various steps of the LC-MS/MS process. The location in the workflow depends on the methods of extraction and analysis. The manual transfer of samples from tubes or wells to other vessels, such as during SPE, can result in a positional sample mix-up. The tracking of individual samples with barcodes and secondary checks is essential to prevent such errors in sample preparation; automation of liquid handling steps is an important tool [98]. If 96-well plates are used and the laboratory has sufficient volume to test multiple plates per day, the location of QCs or calibrators in the 96-well plate layout may indicate plate misidentification in the system’s hardware/software.
Deviations in the steps defined in the SOP must be detailed simultaneously with sample preparation. Procedural errors may also be associated with actions outside sample extraction. For instance, incorrect preparation of a working IS solution and subsequent lack of lot-to-lot verification can lead to an intra-analytical error. Thorough real-time documentation of all processes, including reagent preparation, is essential to root-cause analysis for out-of-specification events [99].
Contemporary LC-MS/MS systems integrate multiple electronic checks. Most monitoring identifies gross errors that result in immediate system shutdown, often accompanied by LC unit beeping, the sound of sudden end of gas output, or the crash of an autosampler arm or needle. Causes, such as acute blockage of the LC tubing, vacuum pressure loss of the MS system, or abrupt power outage, generally do not present symptomatically prior to the terminal insult, but rather occur abruptly and without a warning. The automated nature of LC-MS/MS systems allows for independent operation after acquisition has started, providing for some degree of walk away capability. For any laboratory, an occasional check of the run status is advised as some errors may not have an immediate visual or auditory consequence.
The tracking of intra-analytical QA elements has been recognized as an opportunity for improvement in proteomics [100]. This provides a launchpad for implementation in LC-MS/MS clinical workflows. Data dimensionality is derived from proteomics experiments, but the core components can be readily transcribed to LC-MS/MS assays. Features such as peptide charge state ratios are analogous to qualifier/quantifier transition ratios and provide a framework for executing real-time data evaluation [101]. Attributes of interest to quantitative clinical LC-MS/MS also include results above a threshold (response or concentration), real-time QC result analysis, drift in retention time or peak features, suspected carryover, and alert values for both the system and samples (e.g., LC pressure or insufficient volume, respectively).
Post-acquisition data review
While LC-MS/MS assays are recognized for improved selectivity and sensitivity compared to other technologies, LC-MS/MS offers a perhaps much more important differentiator—the ability to offer proactive QA [102]. Beyond physical/instrument checks (e.g., clot detection) and QC results, alternative quantitative technologies offer little information on the veracity of sample analysis. Only comparisons to external methods or physician feedback from test result-patient presentation disparities can elucidate errors reported by certain analytical methods (e.g., immunoassays). These events are only reactionary, whereas pre-emptive evaluations to detect immunoassay errors (e.g., serial dilutions of all samples or pre-screening for heterophilic antibodies) have been deemed unjustified [103]. In contrast, a well-designed LC-MS/MS assay has multiple layers of data-derived checks ensuring that the result conforms with a quality expectation.
Table 4 lists different components of data review that may be incorporated in a quality review program. Each feature is determined from a batch that includes a standard curve, QCs, blanks, and patient samples. In instances where less frequent calibration and/or QC procedures are utilized, each of these metrics still apply. However, judicious criteria must be implemented to achieve the expectation of quality when error-detecting materials are assessed less frequently.
A benefit exclusive to MS analysis is branching ratios or transition ratios, which are calculated from the peak area relationship of distinct MS/MS acquisitions of a single measurand [104]. These values provide additional evidence of specificity on a per-sample basis. In clinical assays, transition ratios have been applied to small and large molecules [105, 106]. This specificity check has been included in vendor software as well as in in-house solutions to automate this aspect of review [107]. Acceptance criteria for transition ratios are assay-specific but are commonly within 20% of the mean of the calibrators used in the assay [108]. Certain assays may require other criteria, depending on the compound measured [109]. Constraints of the AMR, relative response functions between different transitions, and clinical utility may add context to acceptable transition ratio ranges. Compounds with a relatively abundant quantifier ion and a low collisional dissociation cross-section qualifier ion may require expanded criteria at lower concentrations because of imprecision of integration of the low-abundance qualifier. Similarly, high concentrations may yield a flattening of the dose-response function for a more efficiently detected ion, while a lower-yield product ion is detected in a proportional manner. Here, a broader acceptance range may be appropriate at elevated analyte concentrations. In both instances, the clinical/interpretative implications of a lack of specificity with expanded ratio ranges should be considered.
IS recovery (e.g., the IS peak area of a sample relative to the calibration standards/QCs or entire batch) is a valuable LC-MS/MS QA feature. Not only does it serve as a quantitative normalization function, but it also can identify gross preparative errors or significant ionization suppression or enhancement. IS recovery plots may indicate extreme outliers. Significant scatter of the IS, deviating from that observed in real samples during assay validation, may indicate a need for maintenance of the equipment, retraining of the staff, or evaluation of extraction or method parameters to ensure that IS recovery is consistently stable.
The degree to which IS recovery is preferred in a clinical assay has not been set in guidelines, although common criteria suggest ±50% of the mean of the calibrators and/or QCs in the batch or within a predefined range based on validation data [110]. Excessively broad criteria may be unsuitable for the identification of errors in IS addition during sample preparation. Frequent IS recovery failures may indicate an assay susceptible to ionization suppression or enhancement effects and in such case, returning to method development is a possible course of action.
Acceptable IS recovery ranges should reflect the origins of the IS response deviation in the context of the assay. High IS recovery can result from ionization enhancement, excess injection volume, low volume reconstitution, or under-dilution or over-aliquoting of the IS. In all instances but the last (over-aliquot of IS), the analyte-to-IS peak area ratios will be preserved as long as those responses fall within the linear range of the MS detector. Results may be within the AMR, but the instrument imprecisely detects the signal. When the IS increased in error (over-aliquoting), the back-calculated concentration is underestimated proportional to the degree of added IS. Low IS recovery can result from poor extraction efficiency, ionization suppression, low injection volume, nonspecific binding, excess reconstitution/dilution volumes, adsorptive losses, or a low IS aliquot in sample preparation. When IS addition is the root cause, quantitative values will be higher than normal. In all other cases, fidelity of the data at the low end of the AMR is challenged. Thus, in addition to absolute IS recovery variance criteria, concentration-dependent recovery criteria may help ensure data quality.
Pictorialized chromatographic data review is primarily an exercise in comparison. Calibrators and QCs can be inspected for reproducibility of general features from examples derived from the validation included in the SOP. Those same chromatograms can be contrasted to a recent run for detecting short-term performance drift. Samples can be referenced back to the calibrators and the QCs within the batch for continuity of retention time, peak widths, peak shape, IS recovery, and background/noise (Table 4). Note that this comparison is necessary for each transition (minimally two for each analyte and IS). Additionally, observations of detector blinding/source saturation, partial suppression of a peak due to near-eluting compounds, baseline resolution of high-signal interferences, possible carryover, alert values, and samples that require dilution are all important features of data review [108].
The burden of data review is not trivial. The average human can maintain attention to between two and six components of information in short-term memory [111]. The volume of information provided in LC-MS/MS data greatly exceeds that limit. The capability of LC-MS/MS to near-simultaneously monitor multiple additional data features for determining specificity (second product ion for transition ratios), another “extra” data point for normalizing errors (ISs), and an additional data point for determining the specificity of the IS (another transition ratio) yields an excess of chromatographic and response details. This is a blessing for quality determinations, yet an enormous and perhaps untenable burden on human reviewers.
Tools have been introduced to provide data in comprehensive forms, allowing for more facile and higher-quality determinations that reduce the burden on staff [112, 113]. Features used in the visual identification of errors from chromatographic illustrations can be metricized and computed quickly and reliably, allowing for review of the flawed data instead of all data. Stochastic information obtained from previously acquired batches can be used to provide limits for all of the details usually reserved for visual analysis. For example, IS recovery can be normalized to the mean of calibrators or QCs, classifying samples falling outside the pre-determined acceptance criteria as indicated for review. Concentrations or responses above a threshold can be indicated as liable for carryover in subsequent samples. Samples requiring dilution may be identified for corrective actions. Transition ratios that deviate from that of standards can be designated for follow-up according to the SOP of the assay. All these data features can be interpreted without the review of any chromatograms. This approach to automated data review is not new; the need to reduce the variability in human perception of chromatographic data has been recognized for decades [114].
An automated approach to data review inevitably leads to the possible implementation of immediate release of data from validated platforms. In certain contexts, this could be described as auto-verification [115]. In the model provided by the CLSI, auto-verification includes the analysis of pertinent pre-analytical details (e.g., electronic medical records) in addition to data from the analytical phase, including instrument status and test results. Common approaches to confirming the reliability of auto-verification protocols are performing delta checks (comparison to previous results from the same patient), tracking moving averages, and applying critical concentration cutoffs [116]. Given the data dimensionality of LC-MS/MS, auto-verification for these assays must also include an assessment of the veracity of the results prior to uploading the results into the LIS. All features listed in Table 4 can be queried for outliers in addition to any other assay-specific relevant details [117]. As each of the data components in Table 4 are objectively determined by the software and the data can be constrained by limits, manual data review can be reduced in high-quality assays.
Automated data review requires adequate peak integration by the software provided by the MS vendors or third-party solutions. The determination of integration parameters must be evaluated during method validation, including the effect of modifications to such parameters. Smoothing functions, noise thresholds, integration models, and other factors are generally not modified on a per-sample basis without an explanation for the need and validation of the adjustment. Integration parameters that significantly deviate from the validated assay’s settings can indicate necessary maintenance or a non-rugged assay.
Variability in integration success can be monitored for endogenous analytes that are always expected in a sample. For example, methylmalonic acid cannot be absent in serum; a result <70 nM is considered incompatible with human life [118]. Thus, a retention time and other chromatographic metrics must exist for all analyte and IS peaks in a properly prepared sample. Comparison of these metrics for the peaks in a sample (analyte quantifying and qualifying transitions, and IS quantifying and qualifying transitions) allows for the discovery of inappropriate integrations. It is more difficult when the absence of a molecule is the expected “normal” state, as in illicit drug testing in the general population. In these instances, the stringency of integration parameters must be carefully reviewed to achieve automated data review.
Data review may reveal samples that require additional evaluation and corrective action. Example reasons for re-analysis are provided in Table 5. A frequent determination indicates a concentration requiring dilution. Samples with estimated results above the upper limit of quantitation (ULOQ), samples with insufficient volume, and questionable IS recovery due to matrix effects (ascribed to suppression or enhancement) are common reasons for dilution [108, 119]. An approximation of the necessary dilution can be informed at the pre-analytical stage of analysis for patients with known elevations, such as patients with established inborn errors of metabolism [120]. LC-MS/MS assays do not require same-matrix approaches for the diluent as variations in the analytical recovery due to matrix can be mitigated through the use of the IS. Dilution with authentic matrix may be challenged by endogenous analyte concentrations in the blank material, even after charcoal stripping [121]. Further, neat solvent-based dilutions can afford better recovery of the measurand(s) in both sample preparation and ionization for samples with high concentration(s) of analyte(s) or contaminant(s) [122, 123]. High-purity water is an excellent diluent for LC-MS/MS assays. When solvents are used for dilution, adequate equilibration times for the mixtures and ISs established in validation must be included in the SOP [3].
Dilutions can be performed in three modes. First, and most commonly, an aliquot of the patient sample is combined with a known volume of diluent. The combined solution is mixed and the aliquot for analysis is taken for extraction in the assay, using the same procedure as that used for the calibrators, QCs, and other samples. This process affords consistency in the execution of sample preparation within a batch. In the second dilution approach, the sample is under-aliquoted, whereas all other reagents are used at the appropriate volume. The analyte-to-IS peak area ratio preserves the relative dilution. Table 6 demonstrates a 10-fold change in the analyte-to-IS ratio by reducing the sample volume. This mode removes additional pipetting steps, thus reducing adsorptive loss and possible errors during sample handling [124]. Additionally, this mode of dilution is highly amenable to automated protein precipitation or other solvent-addition-only protocols. It only requires the determination of the change in volume during sample aliquoting [125]. Note that in Table 6, the IS concentration of the final extract has moderately increased; this expected change has to be considered during data review. Third, a dilution can be performed without re-extraction of the sample [126]. This is particularly useful when the sample volume is limited (e.g., neonatal samples) or when additional sample extraction results in undesired delays in reporting. Fundamentally, the process is to achieve a lower ion yield, relying on lower injection volumes and sub-optimal source settings. This is similar to the “over-range-detection-and-correction” functions built into some clinical analyzers [127]. In LC-MS/MS, this relies on a linear calibration curve fit and diluting both analyte and IS responses within the linear detection range of the instrument to achieve quantitative accuracy. In this mode, the precision of IS detection can be challenged by the lower response caused by post-extraction dilution. The range of acceptable dilutions should be evaluated in validation and treated distinctly in data review.
Each batch with diluted samples should include a diluted QC to monitor for errors in the dilution process. Generally, higher-concentration QCs are used if the concentration is appropriate to provide a result within the AMR after dilution of the QC material [128]. The steps taken in dilution of the patient sample must be also applied to the QC to ensure the appropriate processes were followed.
ASSAY MAINTENANCE
LC-MS/MS assay components are consumables. They include pipette tips, QC solutions, and extraction materials as well as chromatography columns, ionization electrodes, MS source heaters and, to a far lesser extent, the instruments themselves. Each constituent will eventually require replacement. The assay volume may also increase beyond the laboratory’s current capacity, requiring the purchase of new equipment. Patient samples should not be tested before the performance of the new material/equipment has been verified. This is referred to as “lot-to-lot verification” or “between-lot verification.” Guidelines and published papers have reported procedures for performing these evaluations [129, 130]. Guidelines recommend that previously analyzed samples be re-assayed using the new material and the results compared [108]. However, this approach may not be appropriate for LC-MS/MS assays. For example, an analytical column has been degrading over multiple days, leading to failure of separation. In comparison with a new column, a previous batch assayed with the degraded column shows a bias in sample results. It may be difficult to ascertain whether that bias is due to the new column performing poorly or whether the old column is the source of error. Additional variables, such as analyte stability, batch-to-batch calibration variance, and even technical staff differences, can affect comparative outcomes. SST injections may be considered to provide excellent evidence for LC-MS/MS column performance, particularly if known isobaric species are included in the SST.
Lot-to-lot variation studies for each material being replaced should be considered for associated risk, variables, and necessary evidence to provide confidence in the new component. Instrument-based materials, such as new mobile phases, columns, or source electrodes, may be accepted by the use of SST and blank injections. Extraction materials, such SPE stationary media or LLE solvents, may require a more thorough evaluation of a new lot. As a thought experiment, the closer a component is to the actual source of the mass spectrometer, the less likely it is to cause issues with individual samples. Issues proximal to the mass spectrometer may cause errors in all samples, including calibrators and QCs, thus rendering the assay “out of control.” In that sense, simplified assessments via SSTs are used in some instances, whereas sample-based verifications are preferred in others, as shown in Table 7.
Perhaps the most critical materials in LC-MS/MS assays are calibration standards used to generate a calibration curve. On a practical level, the entire LC-MS/MS procedure has no intrinsic accuracy requirement, but rather, within-batch precision is the goal [8]. For example, an imaginary assay SOP states that “50 µL of sample, calibrator, QC, or blank shall be aliquoted to each well of a 96-well plate.” If a technician inadvertently aliquots 55 µL for each of the sample types, this will not result in a 10% bias in the data. The calibrators and samples will have been aliquoted at a precise volume within that batch. Assay accuracy is thus derived from the accuracy of the concentration(s) of the measurand(s) in the calibration materials and the precision (relative to treatment of calibration solutions) of each analytical step in the procedure. Further, when isotopically labeled ISs are used, the only requirements are precision of the sample and IS solution addition—such is the power of LC-MS/MS. However, in instances of poor calibration accuracy, indications of failure are limited. These can include requested follow-up testing to resolve discrepancies between test results and patient presentation, inquiries from clinicians, or a notification of a proficiency test result failure.
New calibration materials in the laboratory must be prepared with exceptional care. Close surveillance of expiry or depletion of a current lot of standards allows for adequate time to prepare, subaliquot, verify and, if necessary, correct a newly prepared lot. At least two weeks prior to the conclusion of the current lot’s utility is the minimum time necessary for new standard preparation, especially for tracing and resolving errors determined in verification. More challenging calibration systems (i.e., those with multiple components) benefit from longer lead times.
The collection and analysis of the blank matrix is a critical first step. The exact matrix type, quality, and manufacturer should be determined in validation and published in the assay SOP [131]. In case of deviations from the validation-established matrix due to availability issues or a change in the preferred vendor, the matrix must be re-validated prior to use [6, 8, 9]. This is particularly important in endogenous assays using a depleted or stripped matrix. Distinct vendors may utilize discrete procedures or sampled populations (e.g., gender, age, geographic location of donors) to manufacture stripped matrices [121]. Pre-screening of the blank matrix should include a comparison of the response function of extracted blanks with the current standard lot LLOQ. Any observed response feature of the analyte(s) at the appropriate retention time represents a contamination. An acceptable level of 20% of the LLOQ signal (integrated peak area, not the calculated concentration) is consistent with guidelines [6, 8]. Unacceptable blank matrix contamination can be managed by three means. Accurate assessment of the concentration via standard addition can be used to adjust stock fortification volumes to achieve accuracy of the expected concentrations [132]. Dilution of the contaminated matrix can be performed until contribution to the analyte(s) is no longer observed, although this should be limited to preserve commutability. Lastly, the contaminated matrix can be returned to the vendor, replaced with a new lot, and retested.
Other essential materials include Class A volumetric flasks and pipettes, a calibrated mass balance, and vessels for sub-aliquoting to assay-specific volumes. The latter component infers that bulk calibration materials are prepared, apportioned, and stored at useful volumes (i.e., one aliquot per batch/shift/day as stability allows). This is distinct from freshly preparing calibration solutions with each batch of standards by spiking at the laboratory bench. If an analyte is stable in matrix, long-term storage of bulk preparations decreases labor and removes the possibility of variation/error from repeat calibration preparation.
Stock solutions for fortification can be prepared in-house from lyophilized materials using gravimetry and dissolution. Alternatively, a solution at a known concentration can be purchased and used for fortification. In either case, review of the certificate of analysis is required to capture the impurities/salts and adjust analyte concentration. If the laboratory has access to appropriate equipment, assessment of the concentration of solutions using alternative methods (e.g., spectrophotometry) prior to fortification can provide confidence in the concentration [133]. Spiking of the fortification solutions must follow some recognized rules. First, Class A glassware is preferred over other means of liquid transfer, especially air-displacement pipettes. Air-displacement volumes are calibrated to water; if organic stock solutions are used in standard preparation, there will be a bias in the final concentration due to density differences [134]. Pipettes must be calibrated to the density of the solution utilized or the delivery volume modified to account for different solutions used in preparation. Efficient preparation and prompt subaliquoting and storage is recommended.
The verification of new calibration solutions has no defined path in regulatory guidelines, but method comparison can be a useful framework [135]. Calibration verification is broadly described as experimentally demonstrating that materials of known concentrations, measured as patient samples, produce the expected concentration [136]. For commercially available test systems, calibration materials are validated by the manufacturer prior to release; only verification of those concentrations is required by the laboratory. In contrast, the laboratory is the manufacturer of most LC-MS/MS tests and abbreviated calibration verification experiments may be insufficient to identify errors in standard curve preparation.
Calibration verification for new in-house prepared materials should include assessment of their inaccuracy and imprecision. When possible, a demonstration of trueness by comparison with a certified reference material (CRM) or a reference method procedure is encouraged [108]. Proficiency test (PT) samples with only peer-group mean values can be used in the certification of new standard concentrations, as long as that material is commutable and within expiry. The analysis sequence should be designed to account for LC-MS/MS-specific variables, such as longitudinal calibration drift. A direct correlation of calibration lots can be performed in a single batch, preferably with at least triplicates of the new lot of calibrators. These must be measured as “unknowns” to not influence the curve regression. The precision of the triplicates should be within the expectation for the assay; deviations require re-preparation of the batch or re-analysis. If the mean of the new calibrators falls within the accepted bias of the assay, additional batches can be prepared using patient samples for result comparison. In batch-calibration mode, a current calibration curve at the beginning of the batch and one for the new lot of standards at the end of the batch is acceptable. Two separate standard curve regressions are used to independently calculate the patient concentrations. Bias plotted by sample index can be used to observe any drift in detection within the batch, leading to instrument maintenance and re-analysis. Multiple batches should be performed (n≥3) with samples containing concentrations across the AMR; Deming or Passing–Bablok regression and a Bland–Altman data plot are useful data reduction techniques. The actual choice of regression technique is up to the laboratory, although as suggested by Westgard, acceptance by both models is a valid criterion [137]. The number of samples and the distribution of their results should be rationalized per assay [138]. Efforts should be made to incorporate samples spanning the AMR as is reasonably possible without unduly influencing the regression [139]. Fortification or dilution of samples may be necessary if a high percentage of samples are in the low or high end, respectively. Acceptance criteria are consistent with method comparisons (e.g., Deming slope between 0.9 and 1.1, correlation coefficient >0.98) and Bland–Altman plots should indicate random bias around the unity line. In cases where a minimum bias differs from the state of the art, criteria are narrowed down to match the expectations for allowable bias. Note that intercept review is generally not indicated as interpretation can be muddled by the dynamic range of the assay; intercept interpretation should be done carefully [140]. Any CRMs or PT samples must back-calculate within the tolerances allowed by the assay or the material provider. Values for QCs measured against the new lot of standards should agree with the previously determined mean within the expected variance for each concentration.
New lots of calibration material must be reviewed longitudinally against previous lots [141]. Comparison of the back-calculated bias across multiple lots can indicate a persistent trend (positive or negative). In the case of a constant negative bias, the stock material should be assessed for stability or, in the case of lyophilized materials used consecutively, hydration of the material over time. Multiple positive biases across lots may indicate instability of the previous lot of calibrators used as standards in the comparison. In this case, the re-assessment of calibrator stability is indicated.
In some unfortunate instances, calibration curve verification fails. Root-cause analysis may indicate the source of the issue, but due to time constraints, a new preparation and verification may not be possible. The transformation of calibration curves (modification of theoretical values) should be performed cautiously. Significant deviations from the AMR, particularly at the LLOQ or ULOQ, are not allowed; such changes must be communicated to assay end-users to determine their effect on clinical decisions. Multiple replicates of the new lot should be assessed against known materials to establish the accurate concentration (n≥10). External reference samples (e.g., CRMs, PTs, external quality assessment [EQAS] materials) should be measured in several batches, using independent calibration with the new lot in each batch, to determine the effect of the changes on reported values. Rapid send-out of those calibration materials to a reference laboratory is recommended after confirmation that the materials are commutable with the external method. If transformed calibrators are implemented, the results from real patient samples must be closely monitored for unexpected shifts in the expected values or reference intervals.
The preparation of new lots of QC is similar to that of new standard curve materials in terms of planning and materials. The manufacturing of QCs should be temporally distinct from that of calibrators [141]. Bridging QCs between calibrator lots prevents the possibility of running out of both standards and QCs at the same time. Separate preparation of new standard and new QC lots also affords uninterrupted within-laboratory traceability to the originally validated method. Separate stock solutions should be utilized in calibrator and QC preparation [142]. Distinct stocks can capture errors in the concentration of a material that may be concealed had the calibrator and QC lots been prepared simultaneously from the same stock solution(s).
For each QC, exogenous analytes should be fortified to known concentrations into authentic matrix [6]. Similar to calibration materials, the blank matrix should be verified prior to fortification. When assayed, concentrations should back-calculate to the expected, allowing for imprecision. Deviations in the accuracy of the QCs may indicate a process error (manufacturing) or a calibration error. For non-commercially available exogenous molecules with specific features, collecting patient samples known to be positive for the analyte(s) and assessing the mean is appropriate. These instances include molecular species that are not easily synthesized, such as rare drug conjugates that hydrolyze prior to LC-MS/MS or biologically modified compounds [143].
Endogenous measurand QCs can be prepared by fortification into stripped matrix or generated as a pool of authentic samples. These two types of QCs can also be combined to provide for checks against accuracy (fortified stripped matrix) and matrix effects (authentic pool) [144]. Authentic sample pools can be over-fortified to generate higher concentrations or mixtures of pools used to generate multiple QC ranges [145-147]. Analogous to calibration preparation, materials should be prepared efficiently, subaliquoted to an appropriate volume for use, and stored under SOP-defined conditions. Assay-specific steps, such as mixing time to ensure equilibrium of the fortified samples, should be followed explicitly to prevent variable QC results, especially for analytes with high-affinity binding partners.
The qualification of new QC lots has been described [62]. Qualification studies can consist of preferably 20 replicates across 20 days (batches) or as few as 10 days (batches). Attempting to capture normal laboratory variation is essential. Importantly to LC-MS/MS, variation in calibration curves is as critical as replication of QCs in determining the mean concentration. If an assay is performed on more than one LC-MS/MS system, QC qualification should be performed for each system, with an expectation of consistent results. When the measurand(s) are fortified to a known concentration, estimated results should be near the expected value. In this instance, the theoretical value can be used as the target, requiring fewer replicates in verification. The verification of commercially available QCs is also recommended, even if the manufacturer provides expected target concentrations, though these can be qualified with fewer replicates if desired. All new QC materials, regardless of their origin, should be evaluated for mean and imprecision. Significant changes in the imprecision (variance) of a QC concentration between lots requires investigation. A common cause of shifts in CVs is attributed to a lack of homogeneity. Post-implementation monitoring of QC results may indicate a required shift in the mean of the QC material if insufficient variation is applied in the qualification phase.
System maintenance
A medical laboratory may face challenges related to the deployment of LC-MS/MS assays that are not encountered with other assay technologies. To understand the scope of the LC-MS/MS challenges, analogizing the platform’s use is appropriate. Fundamentally, human blood, urine, or cerebral spinal fluid is chemically manipulated to form an injection solution. A volume representing a fraction of a single drop of water is introduced into a liquid flow operated at pressures near those found in the deep of the ocean (Challenger Deep, Mariana Trench, 1,060–1,140 bar). The sample is carried through tubes that have an internal diameter similar to that of a single thread of hair (25–175 µm) into a particle filter made up of millions of near-perfect spheres that are smaller than many bacteria (1.7–5 µm). The chemicals in the sample separated by these tiny particles are passed through an electrode that operates at several thousands of volts. After exiting the electrode, the sample is exposed to temperatures capable of melting lead (327.5°C) and even plutonium (639.4°C), evaporating nearly all the liquid. Some of the evaporated (gas-phase) molecules can be charged to produce ions, which then pass through multiple electric fields into a vacuum with a pressure comparable to that at roughly 300 km above the Earth’s surface (where space suits are required to support human life). The ions are further energized so that some of them break apart in a process similar to a controlled car wreck (collisional dissociation). The remnants of the ions are further submitted to controlled electric fields after which they are measured, ion by ion, on a small detector. Clearly, LC-MS/MS is a sum of extremes that are the causes of the challenges. Further, platform operation and maintenance are done by clinical laboratory technicians who are responsible for delivering results from the above-described process with minimal downtime. As such, deep knowledge of chromatography, MS, engineering, electronics, physics, biology, and chemistry, as well as a mechanical inclination, are required to troubleshoot all of the possible issues that can arise from LC-MS/MS. Knowledgeable and experienced staff with specialized skills may be difficult to acquire and retain [148]. Appropriate staffing levels to maintain the laboratory’s expectation for performance may be challenging and will be addressed briefly below.
Instrument maintenance can be planned or unplanned. Both chromatographic systems and mass spectrometers have components that require replacement after use. Pump seals, pistons, autosampler needles, tubing, contact surfaces of rotating valves, electrodes, MS tuning values, and filters all have certain lifespans [149]. In general, manufacturers provide recommendations on the replacement frequencies, some of which depend on the number of injections/uses (i.e., valve rotations), while others are temporal. The actual assay performed on a platform also plays a role in the frequency of planned maintenance, as some analyses introduce more contaminants into the flow path or ion optics than others. These contaminants may be of biological origin, such as lipids or proteins. Contaminants of artificial origin, such as manufacturing impurities in SPE media that co-extract with the analytes of interest, may also occur [15].
MS cleaning protocols differ widely from laboratory to laboratory and vendor to vendor. Published manufacturer directions have some similarities in cleaning procedures. Waters, Sciex, Shimadzu, and Agilent all recommend combinations of high-purity water with either methanol/isopropanol or acetonitrile as cleaning solutions [149-151]. Certain protocols recommend the use of detergents such as Alconox or even chlorinated solvents or formic acid at high concentrations. Regardless of the cleaning method used, all procedures recommend adequate rinsing with Type I water and drying of the components before placing them back into service. If nonpolar solvents are utilized, moderate-polarity solvents must be used prior to water rinsing. Solvents may be evaluated for possible contaminants, but logic indicates that higher-purity solvents are less likely to inadvertently foul a system than lower-quality solvents.
The tuning of a mass spectrometer can be a complicated affair. Certain models/systems require significant user interaction to perform mass resolution and mass accuracy adjustments, while in others, these are automated. In either case, knowledge of the effects of tuning errors are critical in maintaining a proper assay. One error that is frequently reported in the literature is the acquisition of ions that are difficult to rationalize because the reported m/z does not match the theoretical m/z based on the atomic constituents. Thyroxine (C15H10I4NO4) measured using MS has been stated to have a measured precursor m/z of 775.9, yet the true mass of a deprotonated precursor is 775.68 [152]. Methylmalonic acid has been measured as a precursor m/z of 116.77, while the true deprotonated precursor m/z is 117.08 [153]. These errors may be due to auto-tune functions executed during initial MS infusion or a deviation in the mass accuracy of the system during method development.
Incorrect m/z values may have consequences. For example, the model molecule phencyclidine (PCP, monoisotopic weight, 243.1987) was measured on an accurately tuned mass spectrometer at the expected [M+H]+ mass value of 244.2, with a product ion of 91.0. The precursor m/z values were offset by 0.2 from the protonated mass, yielding three transitions: 244.0 → 91.0, 244.2 → 91.0, and 244.4 → 91.0. Replicate measurements were performed in the same MS/MS experiment (to resolve the variable of subtle injection volume differences) and the results included the IS at the expected m/z. The data are provided in Table 8, demonstrating a loss of signal for the inappropriate m/z values. Ensuring that the expected values are measured and properly maintaining the mass accuracy of the MS/MS system is a simple exercise in providing optimal system performance.
Misidentification of the appropriate mass or poorly calibrated mass accuracy may increase the likelihood of interfering signals. Most quadrupoles operate with mass resolutions that provide unstable trajectories for compounds more than an integer away from the intended m/z (e.g., 1.0 amu). Measuring the incorrect mass because of an error in the acquisition method or inaccurate MS tuning can lead to undesired ions being detected. Both the loss of signal and the possibility of new interferences can cause distress in an MS/MS laboratory and should be avoided if possible.
TROUBLESHOOTING
Despite best efforts in method development, no assay is guaranteed to be immune from interferences. The very nature of both MS and biology will not allow an assay to be perfectly selective if used on a sufficient number samples [102]. To address this, alternative procedures may be deployed to manage infrequent interferences by leveraging additional chromatographic fidelity. For example, a method for measuring 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (THC-COOH, the primary metabolite of marijuana) using LC-MS/MS was validated in 2017. The assay was validated against all interferences available at the time. In the beginning of 2019, a number of sample results were rejected due to an unknown interferent closely eluting to THC-COOH. An example of this interferent found in a patient sample is shown in Fig. 1. It was determined to be an isobar of THC-COOH, specifically, the delta-8 isomer, which has seen a recent acceleration in abuse rate in the US [154]. To manage such results, an alternative methodology was developed to increase the chromatographic resolution. The original validated gradient and revised gradient are shown in Table 9. Briefly, the modification was to lower the initial %B and lower the pitch of the gradient to achieve a better resolving power, as visualized in Fig. 2. The modification was validated by re-analysis of 70 THC-COOH-only samples from three different batches using the modified gradient, quantifying the results from the original calibration curve, and comparing the reported concentrations using Deming regression. Transition ratios for all three transitions agreed between the original and modified separations. This method is not applicable to all samples. The prevalence of the interferent is such that additional time spent in analysis by reflex injections is less than the time required to assay each sample with the modified method. Future increases in the prevalence of the interferent would require re-evaluation of the practice to possibly incorporate all samples.
PT FAILURES
Proficiency tests and EQASs are critical to the harmonization of assays by ensuring the accuracy of test results, regardless of the technology used [155, 156]. Despite its clear advantages over other platforms, LC-MS/MS assays are not assumed to be perfect and proficiency failures do occur. General reasons for PT failures reportedly are calibration errors, reportable range, instability, component failure, method bias, or indeterminate (of unknown origin) [157]. In addition to these root causes, LC-MS/MS presents distinct sources of PT failure, including variations in matrix effects, lot-to-lot variation of components/reagents, poor specificity, and non-commutability of the PT/EQAS materials [158].
PT/EQAS failures require an investigation of the root cause. This often starts with a review of documentation associated with the test sample, followed by sample handling/reconstitution evaluations, sample preparation error investigations, reagent/calibration record examinations, and finally, instrument maintenance records checks [159]. For LC-MS/MS assays, calibration error is often a cause of inaccurate PT/EQAS results, particularly for a measurand for which a reference method procedure or CRM for multiple laboratories to harmonize to is lacking. As most LC-MS/MS assays are developed in-house, sources of calibration material may differ between labs participating in a PT/EQAS scheme. Preparation methods may differ, such as gravimetry from a lyophilized material in one laboratory versus spiking of a liquid stock solution in another. Different sources of standard stock material may have different qualification procedures, variable quality and purity, and distinct assignments of concentrations, resulting in disagreement between assigned concentration(s) [160-162]. Laboratories running the same analyte may benefit from cross-site trades of calibration materials, QCs, and patient samples at intervals to ensure that neither acute nor longitudinal drift has occurred [163].
A lack of commutability of the test sample can result in PT/EQAS failures, particularly in assays with leveraging extraction modes that are more complex than protein precipitation [164]. Additives intended to maintain stability or inhibit bacterial growth may alter the pH, change the solubility of either the measurand(s) or other species, or introduce unusual ionization features [165, 166]. Experimental approaches to determine non-commutable materials in an assay can include multiple steps. Sample dilution (according to the assay SOP) is viable if the concentration is sufficiently high for the AMR [167]. Post-column infusion comparison of a blank injection, authentic matrix, and a PT/EQAS sample can be informative for abnormal ion suppression [168]. This can be performed for endogenous analytes by extracting the samples without IS and using the IS as an infused analyte, thus resolving any signal contribution from the analyte. Admixing of the PT/EQAS material and true human matrix may also identify sample-based biases [17].
Unacceptable PT/EQAS results should be viewed as an impetus for improvement, but this improvement can only occur with thoughtful and honest root-cause determinations. Regardless of the degree of error, PT failures commonly have multiple or non-obvious sources. Compounding errors, e.g., calibration bias providing some portion of the inaccuracy and imprecision providing the remainder, or seemingly simple procedural deviations, such as not adequately mixing reagents, can be difficult to detect without systematic investigations [169].
STAFFING
Of all the resources required to operate an LC-MS/MS laboratory, human capital may be the most essential. The skill sets held by excellent LC-MS/MS technicians are difficult to train and, once trained, become highly valuable to competitive industries (e.g., pharmaceutical bio-analysis) [170]. Laboratory generalists may provide satisfactory technical benchwork, but LC-MS/MS specialists are necessary to perform the complex troubleshooting and maintenance required. Laboratories with sufficient numbers of platforms (i.e., >10) often benefit from internalization of primary service functions [171]. For laboratories with smaller LC-MS/MS footprints, external service contracts are essential to operations, but it should be recognized that facilities historically supported by external services (e.g., research or academia) generally have lower service needs than clinical laboratories [172]. Hospital laboratories and reference facilities may operate continually, even during holidays. Service contracts can be structured to align with the laboratory’s expectations for maximum allowable downtime. Alternatively, redundancy in platforms can be instituted by cross-validation of assays on backup systems, if the volumes of both samples and available instruments allow for extension of the service timelines.
Staff acquisition and retention rely on several factors. Training features highly in both aspects [173]. Extensive training and experience may be required for satisfactory platform performance and should be considered a priority by the responsible management [174]. This can be challenging as general qualifications for clinical laboratory technicians seldom address MS methods; vocational training is the most common in the LC-MS/MS industry. Professional organizations, such as the American Association of Clinical Chemistry, offer online programs for top-level instruction in LC-MS/MS [175]. These lack specific details on instrument repair or maintenance; such particulars can be learned from instrument manufacturers or by spending adequate time with service engineers. Retaining trained staff is a balance of management responsibilities, including providing an appropriate work environment and adequate compensation and growth opportunities [173, 176].
CONCLUSIONS
This review intended to capture the lifecycle of LC-MS/MS clinical assays after method development, from validation through to the management of operationalization. Despite sincere efforts to summarize sufficient information and experience of the laboratory processes, this review is not all-encompassing. The diversity of science fields underpinning LC-MS/MS, variation in vendors, and enormous complexity of the target population indicate that a truly comprehensive review is nearly impossible. Each laboratory will experience challenges that are not discussed herein, but hopefully, the content is sufficient to provide experimental paths for solution elucidation.
The potential of LC-MS/MS in clinical testing is enormous. The direct measurement of chemical species with high sensitivity while also providing absolute analytical normalization and adding an integrated specificity assessment are of immense benefit to patients. These advantages overcome many liabilities of other technologies, particularly in terms of selectivity, and offer a means to provide quality data in the age of evidence-based medicine. Hopefully, with ideas derived from this manuscript, continuous growth of the skill set of laboratory staff, and a never-ending aim for better quality, the promising potential of LC-MS/MS can be fulfilled.
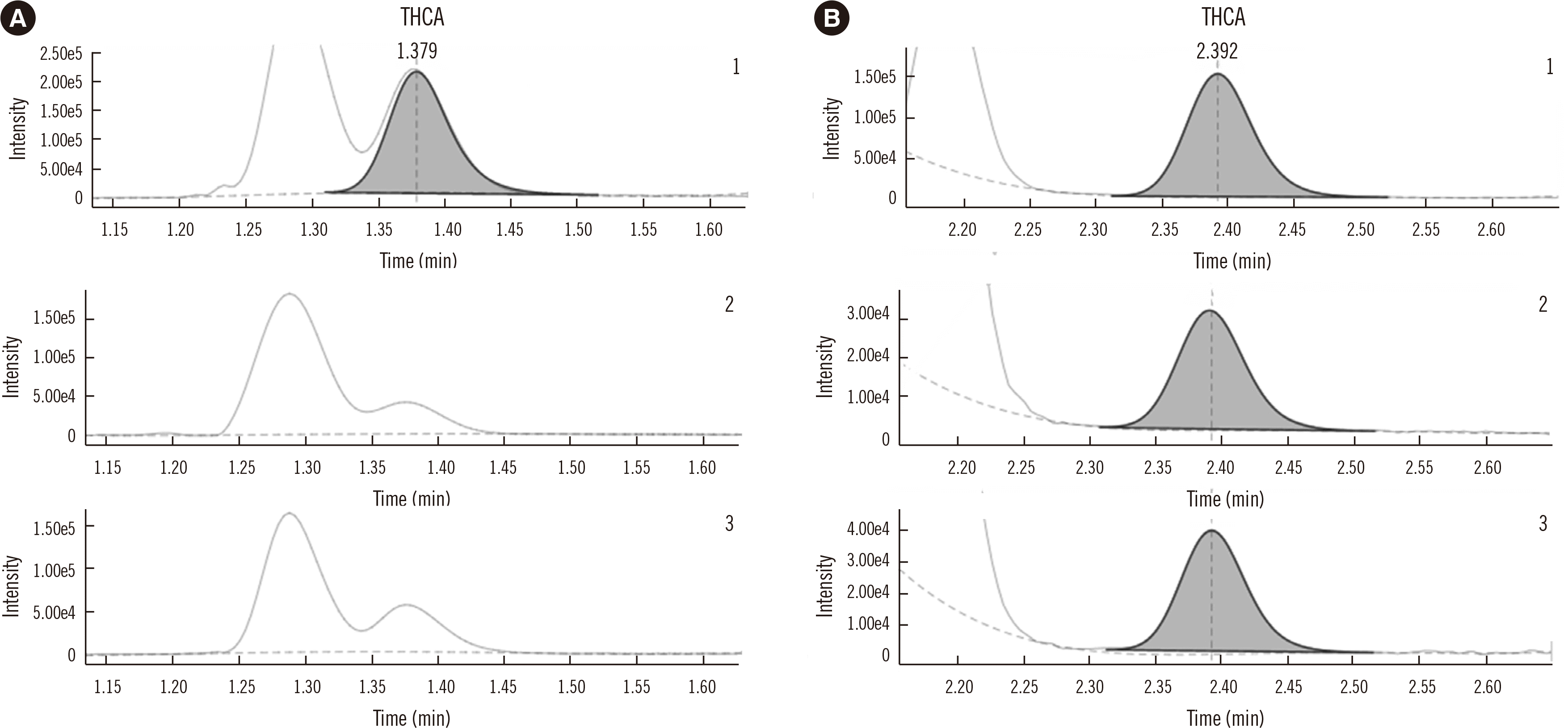
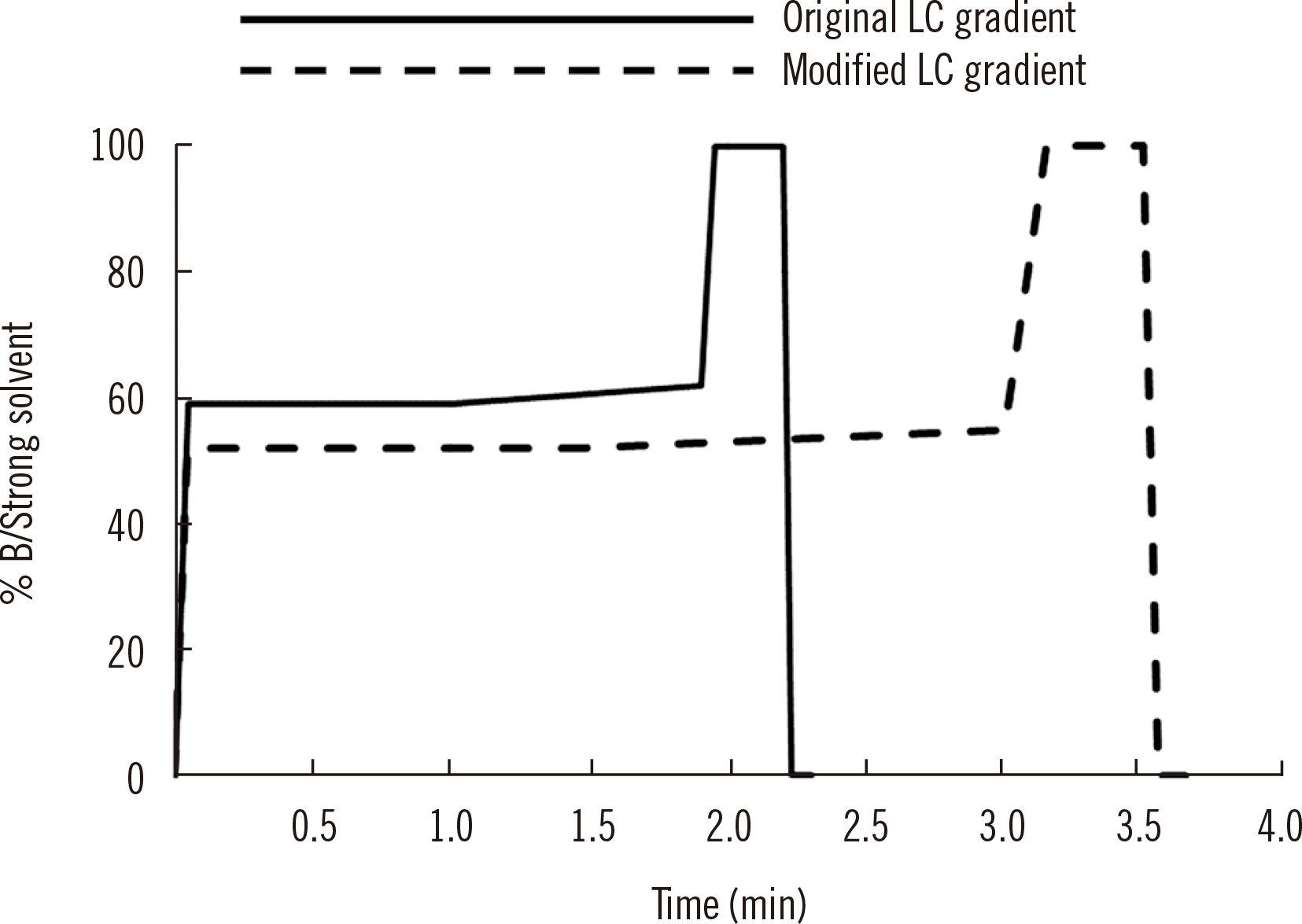
 XML Download
XML Download