

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Inherited bone marrow failure syndrome (IBMFS) is a clinically heterogeneous disorder characterized by significant hematological cytopenia of one or more hematopoietic cell lineages [1]. Different forms of IBMFS can present with or without numerous associated physical abnormalities and a spectrum of pathological findings involving multiple organ systems [2]. The genetic etiology of IBMFS includes germline mutations affecting several key biological processes, such as DNA repair, telomere biology, and ribosome biogenesis, which can lead to four major syndromes: Fanconi anemia (FA), dyskeratosis congenita (DC), Diamond- Blackfan anemia (DBA), and Shwachman-Diamond syndrome (SDS) [3].
The differential diagnosis of inherited bone marrow failure (BMF) from acquired BMF has important clinical implications. Identification of IBMFS prior to hematopoietic stem cell transplantation (HSCT) is important as it allows for the modification of HSCT conditioning and the exclusion of family donors with the same inherited mutation [4]. FA, DC, and DBA are associated with an increased risk of solid tumors and hematopoietic malignancies. Cancer risk was found to be most common in patients with FA, followed by those with DC and DBA, while SDS had a low incidence of cancer [5].
Germline mutations have been identified in approximately 95% of patients with FA and SDS and 70% and 50% of patients with DC and DBA, respectively [3]. Some syndromes, such as FA with autosomal recessive (AR) inheritance, typically develop early in life with high penetrance, whereas others, including a subset of DC with autosomal dominant (AD) inheritance, show variable penetrance and develop in adolescence or adulthood [2]. In addition, recent advances in genomic approaches have revealed previously unrecognized etiologies, many of which occur in adulthood and exhibit less pronounced clinical phenotypes. Here, we review the clinical and genetic features of the major forms of IBMFS and discuss their molecular genetic diagnosis.
CLINICAL AND GENOMIC CHARACTERISTICS
Because of the broad heterogeneity of IBMFS, it is imperative to the diagnosis that we understand the specific characteristics of the different forms of this disease. In this section, we will take an in-depth look at the clinical and genomic characteristics of each of the four major forms of IBMFS.
Fanconi anemia
FA is a rare AR disease [although X-linked recessive (XLR; FANCB mutation) and AD (RAD51 (FANCR) mutation) forms have also been reported] and is characterized by congenital growth abnormalities, bone marrow failure, and a predisposition to cancer. Although patients with FA are rare (1–5 per million), heterozygous carriers are present at a much higher frequency (1/300) [6].
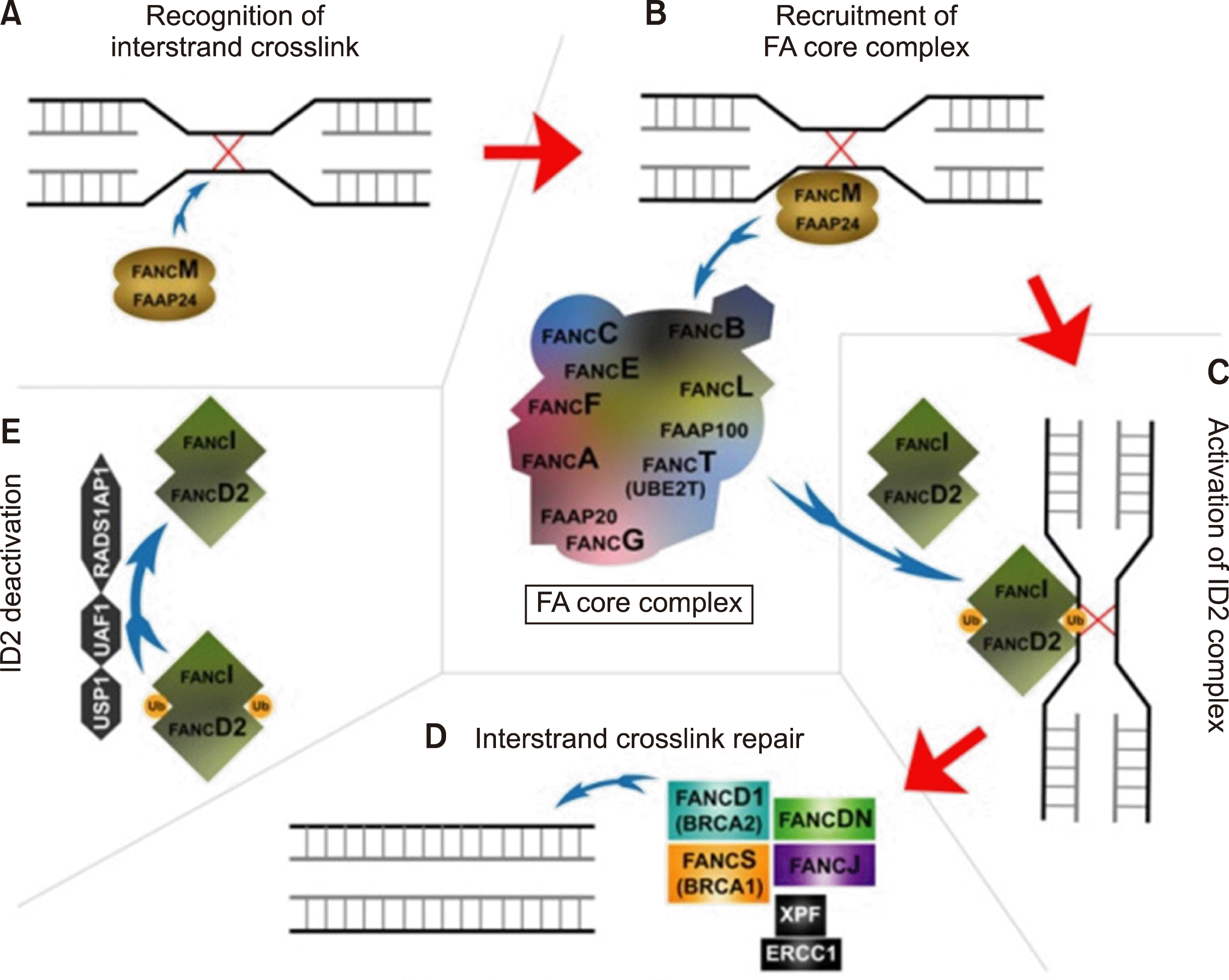
The FA pathway is a fundamental DNA repair pathway that both recognizes DNA damage and modulates DNA damage responses (Fig. 1, Table 1) [7]. This pathway begins with the recognition of interstrand crosslinks between DNA strands by FANCM and FAAP24. FANCM and FAAP24 recruit the multi-subunit FA core complex to the site of interstrand crosslinking, which then activates the FANCI‐FANCD2 (ID2) complex through monoubiquitination (Ub). The activated ID2 complex recruits several different proteins, including FANCD1 and FANCS, which excise and replace the interstrand crosslink. Once the interstrand crosslink is repaired, the pathway is inactivated by the deubiquitination of the ID2 complex.
Twenty-two genes are known to be involved in the FA pathway, and mutations in any one of these genes may cause FA. For FA genes with AR inheritance, biallelic inactivation is necessary to cause FA. However, monoallelic inactivation of some of FA genes, such as FANCD1 (also known as BRCA2) and FANCS (BRCA1), leads to a predisposition for adult-onset cancer [8].
Loss of function of the FA pathway resulting from mutations in the FA gene causes cells to be hypersensitive to DNA interstrand crosslinking agents, such as mitomycin C (MMC), cisplatin, and diepoxybutane (DEB), which subsequently increases chromosomal breakage [8]. Clinically, the accumulation of chromosomal breaks may enable the diagnosis of FA, even without classical physical symptoms, and increased chromosomal breakage predisposes FA patients to cancer [6]. Although most FA-related genes have been identified to date, a small subset of FA patients does not have mutations within known FA genes; whole-exome or whole-genome sequencing will likely be required to identify their genetic defects [8].
Cytopenia is the most common FA presentation. Hematological manifestations include aplastic anemia, myelodysplastic syndrome, and leukemia. The median age at diagnosis of patients with FA is 7 years; however, FA can also be diagnosed in patients over 50 years of age. Patients with FA are at an extremely high risk of developing cancer at an early age; the most common cancers associated with FA are acute myeloid leukemia and squamous cell carcinomas of the head, neck, and female genitalia [9]. The physical phenotypes of patients with FA are extremely heterogeneous and can affect multiple organ systems. Fiesco-Roa et al. [9] evaluated genotype-phenotype correlations in published cases of FA and found that 79% of patients had at least one physical abnormality. The most frequent abnormalities were short stature, upper-limb abnormalities (radial ray), abnormal skin pigmentation, renal malformations, and microcephaly. Additionally, patients with biallelic or hemizygous null mutations had a higher proportion of at least one abnormality than those with missense mutations (92% vs. 75%).
Dyskeratosis congenita and related telomere biology disorders
Telomere biology disorders (TBDs), including DC, are caused by germline mutations in telomere genes that result in very short telomeres. Telomeres are DNA-protein structures that protect chromosome ends from degradation and fusion and are essential for the maintenance of genomic integrity [10]. When telomeres become critically short, chromosome ends are recognized as DNA double-stranded breaks and DNA damage response pathways are activated, leading to apoptosis and senescence [11].
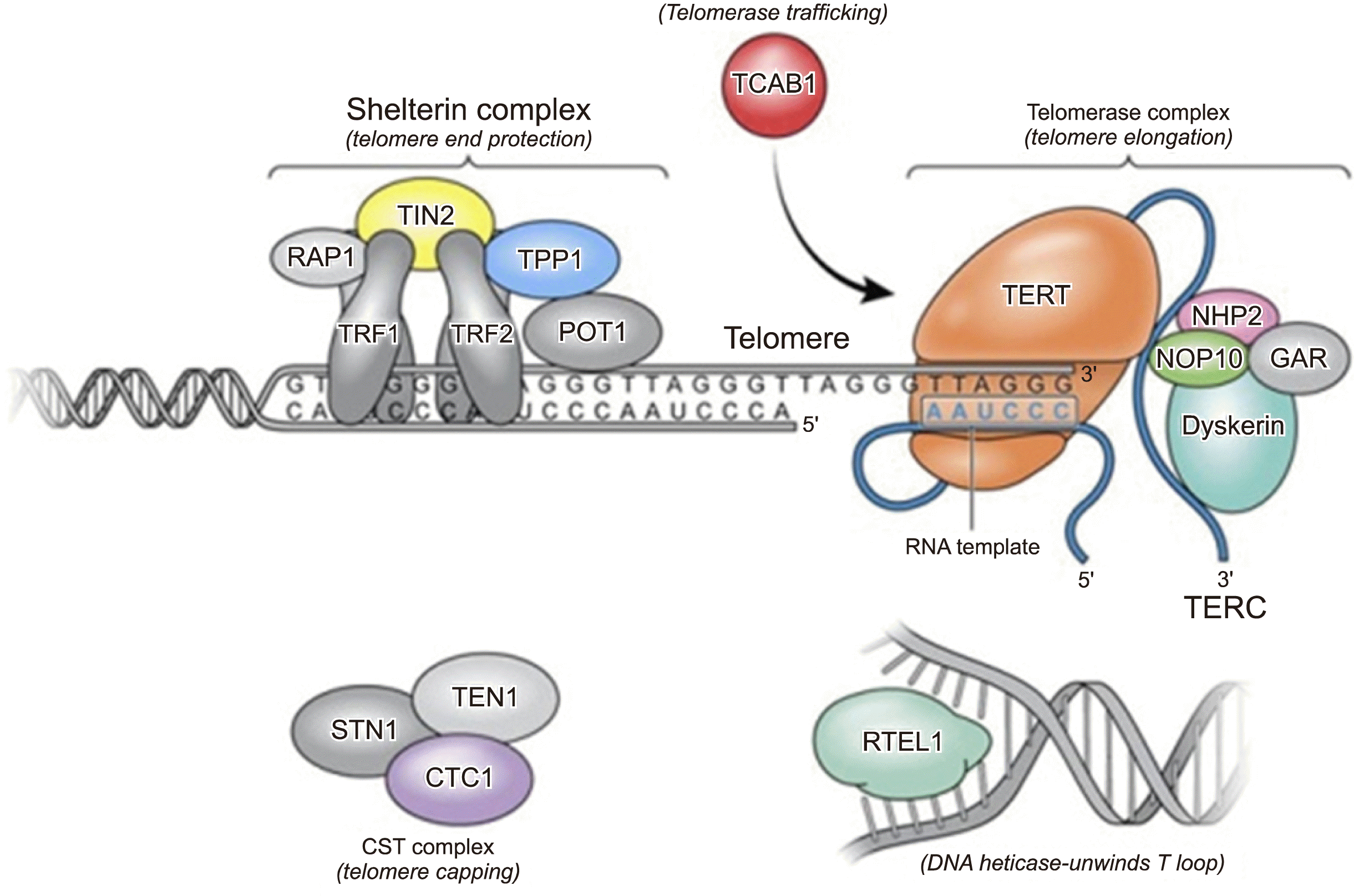
Telomeres are composed of thousands of TTAGGG repeats localized at the ends of linear chromosomes (Fig. 2) [11]. Telomeres are coated with shelterin, a 6-protein complex (comprised of TRF1, TRF2, TIN2, POT1, TPP1, and RAP1 proteins) with multiple roles in maintaining telomere length homeostasis, preventing DNA damage response activation, and recruiting the telomerase complex. The 3' end of the telomeric leading-strand ends with a single-stranded overhang, which invades the double-stranded telomeric helix to form the T-loop. The T-loop is unwound by the DNA helicase RTEL1 for telomere elongation. Telomere elongation is accomplished by the ribonucleoprotein telomerase, which is composed of an RNA template, TERC, reverse transcriptase, TERT, and a 4-protein scaffold of dyskerin, NOP10, NHP2, and GAR. TCAB1 ensures trafficking of the telomerase complex to telomeric ends.
To date, at least 14 telomere genes have been found to be responsible for DC and related TBDs, and 50–60% of patients with DC and related TBDs have mutations in one of six telomere genes: DKC1, TERC, TERT, NOP10, NHP2 or TINF2 [12]. In addition, DC exhibits variable inheritance patterns depending on the genes involved. The classical phenotype of DC consists of the mucocutaneous triad of dysplasia (nail, oral leukoplakia, and reticular skin pigmentation), and these patients are at a very high risk of bone marrow failure, cancer, and pulmonary fibrosis [12].
Patients with DC and related TBDs exhibit variable genetic penetrance and expressivity as well as a broad phenotypic spectrum [13, 14]. Classic DC, Hoyeraal-Hreidarsson syndrome (a severe form of DC with cerebellar hypoplasia), Revesz syndrome (DC with exudative retinopathy), and Coats plus syndrome occur in childhood. Conversely, isolated aplastic anemia, pulmonary fibrosis, and liver disease occur predominantly in adults [13, 15]. Childhood-onset disease occurs with high penetrance, severe clinical symptoms, and specific organ involvement [11]. Hoyeraal-Hreidarsson syndrome and Revesz syndrome are more likely to occur when there are mutations in genes with XLR or AR inheritance and a heterozygous TINF2 mutation [13]. In contrast, heterozygous mutations in TERT, TERC, RTEL1, or PARN are more likely to occur in adults with isolated diseases, such as aplastic anemia and pulmonary fibrosis. Clinical manifestations of adult-onset disease are often milder than in children, and the mucocutaneous triad and other physical abnormalities are infrequently observed [11]. Additionally, in patients with AD mutations, particularly in TERC, TERT, and TINF2, progressively shorter telomeres are inherited from generation to generation, resulting in genetic anticipation in which disease severity increases with successive generations [12, 13].
Diamond-Blackfan anemia
DBA was the first identified human ribosomopathy and is characterized by congenital erythroid hypoplasia [16]. DBA typically presents with erythroid aplasia in the first year of life; approximately 95% of DBA patients are diagnosed before 2 years of age, and 99% are diagnosed before 5 years of age. However, DBA rarely occur in adulthood [17]. In patients with DBA, anemia typically presents as macrocytosis and reticulocytopenia, while neutrophil and platelet counts are usually normal [16]. The bone marrow is typically normocellular; however, erythroid precursors are rarely observed. Additionally, erythroid colony-forming units (CFU-E) are severely reduced and erythrocyte adenosine deaminase levels are often elevated. Approximately 50% of DBA patients have physical abnormalities, such as craniofacial abnormalities (including cleft lip or palate), thumb abnormalities, cardiac malformations, and short stature. Their risks of cancers and myelodysplastic syndrome are elevated, and the cumulative incidence of malignancy is approximately 20% by 46 years of age. Spontaneous remission has been reported in 20% of patients by the age of 25 [17].
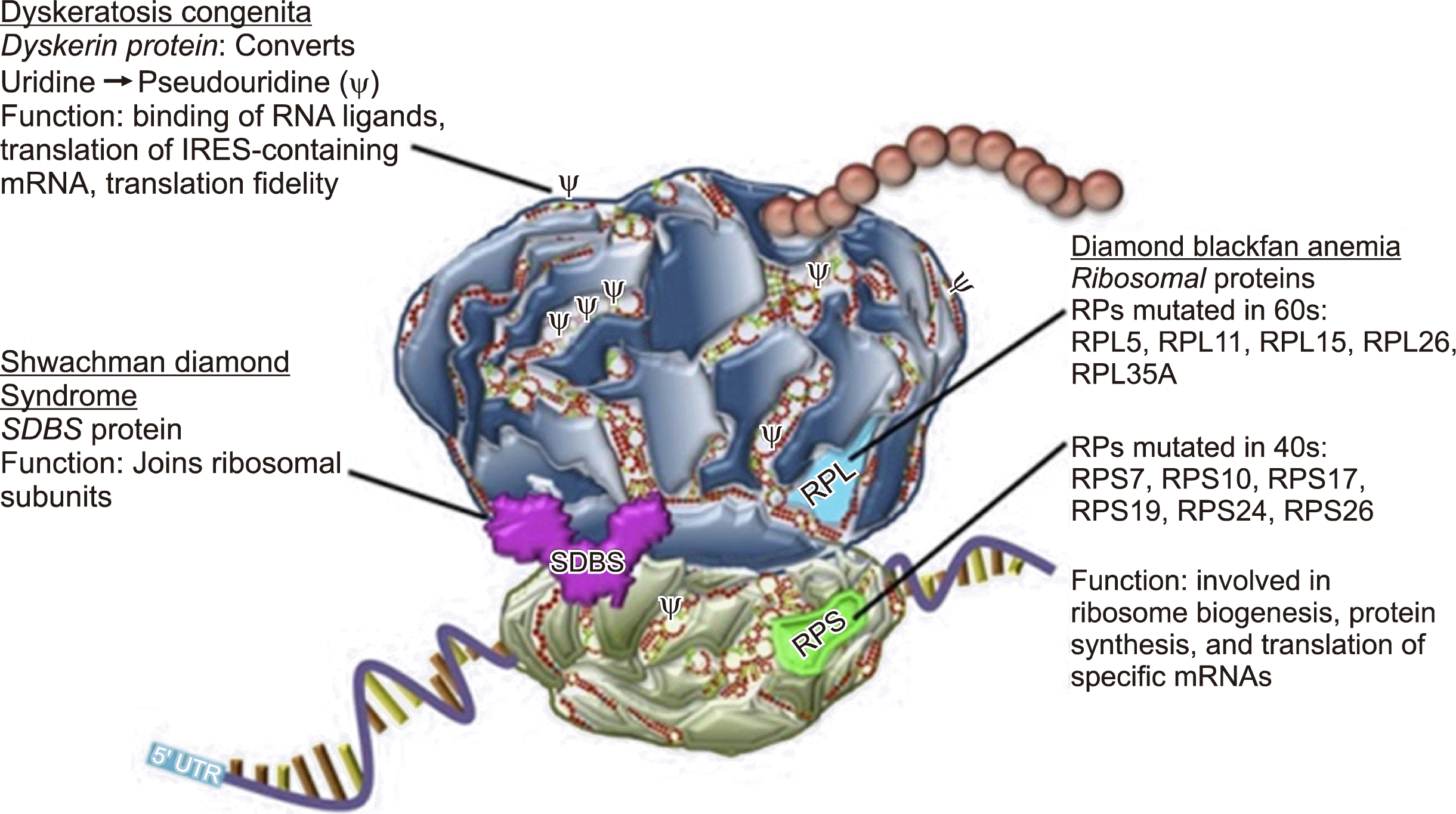
Ribosome biogenesis is a complex, highly regulated process. Eukaryotic ribosomes are comprised of two subunits, the small 40S subunit and the large 60S subunit, which join together to form the active 80S ribosome (Fig. 3). Ribosomes contain four structural ribosomal RNAs (rRNAs). The 40S subunit contains the 18S rRNA, and the large 60S subunit contains the 28S, 5.8S, and 5S rRNAs; these rRNAs are complexed with approximately 79 ribosomal proteins in eukaryotic ribosomes [17]. To date, heterozygous mutations in 18 ribosomal protein genes have been identified in DBA patients.
The loss of a ribosomal protein preferentially affects the maturation of the ribosomal subunit containing that protein and ultimately reduces the number of functional 80S ribosomes. Mutations in ribosomal protein genes account for 60–70% of patients with DBA. RPS19 is the most frequently mutated gene in DBA and missense mutations are the most common mutations in RPS19. In contrast, nonsense mutations, small deletions or insertions, and splice-site mutations are common in RPL5 and RPL11. Large deletions, mostly in the RPS17, RPL35A, and RPS19 genes, have been identified in 20% of patients with DBA; the detection of such deletions requires the use of techniques such as quantitative polymerase chain reaction, multiplex ligation-dependent probe amplification (MLPA), and chromosomal microarray. Ribosomal protein gene mutations may be inherited in an AD pattern or arise de novo. XLR mutations in the transcription factor GATA1, which is essential for erythropoiesis, and in TSR24 have also been associated with DBA-like phenotypes. Interestingly, approximately 35% of patients with DBA remain genetically undefined [17, 18]. Ulirsch et al. [19] reported the results of whole-exome sequencing in a cohort of 472 patients with a clinical diagnosis of DBA. In this study, the authors identified relevant rare and predicted damaging mutations in 78% of individuals. The majority of the mutations were singletons, absent from population databases, predicted to cause loss of function, and located in previously reported ribosomal protein genes [19].
The penetrance of DBA is often incomplete. Patients with DBA exhibit varying clinical severities, ranging from silent phenotypes without anemia to mild and severe DBA phenotypes with anemia. Patients with a silent phenotype are still at increased risk for solid tumors and hematological events, such as late-onset anemia or hematological malignancies [16]. In terms of genotype-phenotype correlations, DBA patients carrying a mutation in the RPS19 gene exhibited fewer malformations than other patients. In contrast, patients carrying a mutation in the RPL5 gene exhibit malformations that may include cleft palate and cardiac defects with multiple complications. A triphalangeal thumb is a common phenotype in DBA patients harboring a mutation in the RPL11 gene, and congenital cardiac defects have been associated with mutations in the RPS24 gene [16].
Shwachman-Diamond syndrome
SDS is a multiorgan disease mainly characterized by bone marrow failure, skeletal abnormalities (including metaphyseal chondrodysplasia, rib cage dysplasia, and osteopenia), exocrine pancreatic insufficiency, poor growth, cognitive impairment, and predisposition to hematological malignancies [20, 21]. Patients typically present with neutropenia and steatorrhea early in life. Although neutropenia is the most common hematological abnormality among SDS patients, anemia and thrombocytopenia are also common. A subset of patients may exhibit atypical or cryptic clinical presentations, and some may develop severe aplastic anemia. The bone marrow of SDS patients is typically mildly dysplastic and hypocellular, and immunologic abnormalities in B- and T-cell numbers and function have been reported, as well as a variety of congenital anomalies affecting skeletal, neurocognitive, endocrine, and cardiac systems [17]. 15–20% of patients with SDS present with myelodysplastic syndrome with a high risk of acute myeloid leukemia transformation [22]. Although early onset solid tumors have been described in patients diagnosed with SDS, the data are insufficient to determine whether patients with SDS are at an increased risk of solid tumors.
Although SDS is a genetically heterogeneous disorder, it is caused by mutations that target a common pathway involved in the maturation of the large ribosomal subunit [21]. SDS is primarily caused by mutations in the Shwachman-Bodian-Diamond Syndrome (SBDS) gene, although three other genes have also been associated with an SDS-like phenotype: DnaJ heat shock protein family member C21 (DNAJC21), GTPase elongation factor-like 1 (EFL1), and signal recognition particle 54 (SRP54). SBDS, DNAJC21, EFL1, and SRP54 are all involved in ribosome biogenesis. SBDS, through direct interaction with EFL1, promotes the release of eukaryotic initiation factor 6 (eIF6) during ribosome maturation; DNAJC21 stabilizes the 80S ribosome; and SRP54 facilitates protein trafficking [22]. Recently, Koh et al. [23] discovered a de novo heterozygous EIF6 mutation using exome sequencing in a 6-year-old boy who presented with pancytopenia (during the neonatal period), liver transaminitis with hepatosplenomegaly, developmental delay, and pancreatic insufficiency, suggesting that a heterozygous mutation in the EIF6 gene may cause an SDS-like phenotype. Normally, eIF6 inhibits ribosomal maturation; its removal during the late stages of ribosomal maturation by SBDS and EFL1 proteins allows the 40S subunit binds to the 60S subunit to generate a functional 80S ribosome.
SDS caused by SBDS, DNAJC21, and EFL1 mutations is inherited in an AR manner. Approximately 90% of patients with clinical features of SDS harbor biallelic SBDS mutations. Interestingly, most SBDS mutations correspond to sequences found in an adjacent, highly homologous pseudogene and disrupt protein production; such mutations likely result from gene conversion [17].
CHALLENGES AND CONSIDERATIONS OF GENETIC DIAGNOSIS
Because IBMFS is a disease that affects hematopoietic cells, there are cases where the diagnosis is challenging when genetic testing is performed using peripheral blood. In this section, we will describe the points to be aware of when diagnosing IBMFS.
Somatic genetic rescue
In Mendelian disorders, cells in which an in vivo somatic genetic event has occurred that partially or totally offsets the effect of the pathogenic germline mutation may gain a selective advantage over non-modified cells; this is known as somatic genetic rescue (SGR) [20, 24]. Unlike somatic reversion, which specifically involves the genetic reversion of the mutated allele to wild type (also known as a ‘back mutation’) and is thus referred to as ‘natural gene therapy’, SGR can result from any type of mutation directly or indirectly that affects the germline-mutated gene [24, 25]. Somatically modified clones with a fitness advantage generate somatic mosaicism and reach detectable levels when they expand, differentiate, migrate, or survive better than the unmodified cells in the body. Because germline mutations in IBMFS-related genes primarily reduce the fitness of hematopoietic cells, somatic mosaicism in patients with IBMFS predominantly affects cells via two distinct mechanisms with contrasting effects [26]: 1) an acquired variation can improve cell fitness towards baseline levels, thereby rescuing a deleterious phenotype; or 2) somatic mosaicism may provide a fitness advantage that results in malignant transformation. SGR has been molecularly characterized in over 30 distinct inherited hematological disorders, including FA, DC, DBA, and SDS. Direct SGR corresponds to spontaneous genetic modification of the germline mutated gene and can be mediated by several mechanisms, including back mutation, interstitial deletions, chromosomal deletions, intragenic recombination, copy-neutral loss of heterozygosity, promoter-activating mutations, and chromothripsis [24]. Indirect SGR corresponds to a genetic modification that does not directly affect the germline-mutated gene but influences another genetic entity that is involved in the pathway affected by the germline mutation. The in vivo occurrence of SGR events in cells of the hematopoietic system generates somatic mosaicism and thus has important diagnostic, therapeutic, and clinical consequences.
When somatic reversion occurs in hematopoietic cells, the diagnosis of IBMFS is challenging [1]. Somatic reversion leading to the correction of pathogenic alleles in lymphocytes has been reported in FA and should be considered in patients with high clinical suspicion of FA but normal lymphocyte chromosome breakage. Because blood is usually used for genetic testing, it is especially important to differentiate somatic from germline genetic mutations in hematopoietic system diseases. In this context, the determination of a germline genetic mutation as disease-associated is facilitated by testing family members and using an alternative source of non-hematopoietic cell DNA, such as skin fibroblasts [1].
Genetic diagnosis
Cultured skin fibroblasts are the preferred tissue for germline mutation testing to diagnose IBMFS in patients with hematological malignancies. A 3-mm skin-punch biopsy or skin ellipse taken at the site of bone marrow sampling or during implantation of a venous device can provide a high quantity and quality of germline DNA. Cultures of fibroblasts can take 3–6 weeks until sufficient DNA for testing is available; however, DNA from the epithelial cells of hair follicles is readily available for time-sensitive studies. Buccal swabs, saliva, or DNA obtained directly from skin biopsies without culture are frequently contaminated with hematopoietic cells and should be avoided [27, 28].
Given the phenotypic overlap, atypical presentation, and genetic heterogeneity of IBMFS, next-generation sequencing (NGS)-based gene panel testing is often preferred, as it offers the ability to analyze multiple genes simultaneously and is cost-effective [27, 29]. However, because NGS-based testing may not capture all exonic and intronic alterations or genomic regions of interest, Sanger sequencing is applied when clinical suspicion of a defined predisposition syndrome is high. In addition, the selection of an appropriate testing method should consider the expected mutation type and molecular genetic mechanism. Overall, whole-exome sequencing provides a powerful, unbiased approach for elucidating the complex biology underlying rare forms of IBMFS by identifying novel genetic causes.
CONCLUSION
An accurate and timely diagnosis of IBMFS is essential for guiding clinical management. However, distinguishing inherited from acquired BMF still faces significant clinical challenges due to overlapping and atypical clinical manifestations. Although some patients show characteristic clinical features of the disease, variable penetrance depending on the underlying genetic mutation contributes to difficulties in diagnosis. Therefore, for the diagnosis of IBMFS, it is important to understand the disease, identify suspect genes, and perform targeted molecular genetic testing. NGS-based gene panel testing or whole exome sequencing, which are currently performed in both clinical and research settings, will help to elucidate the genetic causes and mechanisms governing the pathogenesis of this genetically heterogeneous disease.
 XML Download
XML Download