

 PDF
PDF Citation
Citation Print
Print


THE STORY OF ACUTE MYELOID LEUKEMIA STEM CELL BEGAN
The essential in vivo-experimental methodology to study leukemic stem cells (LSCs) was developed in the early 1960s by McCulloch and Till [1]. They observed that a small subset of mouse malignant hematopoietic cells could proliferate extensively in vitro and in vivo [2, 3]. The looming notion of LSCs became a proven concept with firm certainty in a seminal paper published in Nature in 1994 by Lapidot and Dick [4]. In this study, only CD34+CD38- human primary acute myeloid leukemia (AML) cells were shown to be able to repopulate in severe combined immunodeficient (SCID) mice. They also calculated the frequency of the SCID leukemia-initiating cell engraftment unit as one LSC in 1.2–5.3×105 leukemic cells by limiting dilution analysis from mouse transplant experiment using a patient sample diagnosed as AML-M1. Since this report, AML has been understood to develop into AML blasts from a small subgroup of acute myeloid leukemia stem cells (AMLSCs) present at the hierarchical apex. Subsequent studies further characterized the immuno-phenotype of AMLSCs based on their surface marker expression. CD33 and CD45RA are highly expressed [5], while CD90/THY-1 is negative in LSCs, in contrast to normal hematopoietic stem cells (HSCs) [6]. CD34+CD38-CD90- AML cells have been proposed as AMLSCs capable of initiating AML in SCID mice. Since the discovery of AMLSCs, the general concept of LSCs has expanded to cancer stem cells in solid tumors [7, 8].
WHAT WE KNOW ABOUT THE CELL OF ORIGIN OF AML?
Based on the fact that the unlimited self-renewal capacity in normal hematopoiesis is limited to HSCs and lost in progenitor cells and the discovery that AMLSCs belong to the CD34+CD38-cell population, AMLSCs were hypothesized to originate from HSCs. This hypothesis was strongly supported by the observation that CD34+CD38- AML cells could transfer disease to SCID mice, whereas CD34+CD38+ AML cells could not [4]. In AML, the cell of origin has been largely studied based on the delicate discrimination of leukemic cell populations harboring AML-specific genetic mutations by multi-color flow cytometric analysis and identification of cell populations with LSC capacity validated by long-term culture-initiating cell assay in vitro and/or by immunodeficient mouse transplantation assay in vivo. Although immunophenotype of AMLSC is CD34+CD38, other details of the additional expression markers do not exactly match those of normal HSC. For example, AMLSCs lack CD90 expression, in contrast to HSCs [6]. AMLSCs appear to have progenitor cell immunophenotypes, such as lymphoid-primed multipotent progenitors (LMPPs) and granulocyte-monocyte progenitors (GMPs), rather than HSCs. Goardon et al. [9] proposed the coexistence of two LMPP-like and GMP-like AMLSCs in primary AML samples.
A previous study using a powerful leukemic driver MLL fusion proved that not only HSCs but also committed myeloid progenitors could be the cell of origin of AML in mice [10]. In this study, isolated stem cells and myeloid progenitor populations were efficiently immortalized in vitro, resulting in rapid onset of AML with similar latencies following transplantation in vivo. Regardless of the initiating cells, leukemias generated in mice displayed similarclinical features of characteristic maturation arrest, putatively at a monopotent monocytic progenitor stage. This seminal study revealed that AML arises from HSCs present at the top of the hierarchy as well as committed myeloid progenitors and might present AML with similar immunophenotypes. Some genetic mutations recurrently detected in AML, such as MLL fusion, can transform mutated hematopoietic stem and progenitor cells (HSPCs) into leukemia-initiating cells through the acquisition of self-renewal capacity and immortality. A previous study investigating the origin of acute lymphoblastic leukemia with ETV-RUNX1 fusion, p210 BCR-ABL1 fusion, and p190 BCR-ABL1 fusion found that ETV-RUNX1 and p190 BCR-ABL1 fusion were detected in B-cell progenitor populations, while p210 BCR-ABL1 fusion was detected in the HSC population, which suggested that the self-renewal capacity of the two fusions might differ [11], providing a glimpse of the diverse AML-inducing capabilities of different genetic mutations. However, the impact of each combination of different genetic mutations on the leukemia initiation process from HSPCs could not be deciphered.
AMLSC MODEL WAS UPDATED TO A STAGED LEUKEMOGENESIS MODEL
In rare cases, a single gene fusion of MLL rearrangement with various partners, which is thought to be produced by an insult of topoisomerase 2 poisons, is capable of initiating leukemia even in the fetus [12, 13]. However, most AML cases have multiple gene mutations at the time of initial diagnosis [14, 15]. Advances in next-generation sequencing (NGS) technologies have revealed that gene mutations accumulate in blood cells with aging, even in normal people without a disease phenotype, and this type of mutation is known as clonal hematopoiesis of indeterminate potential (CHIP) [16]. Individuals with CHIP are considered a high-risk group for the development of hematologic malignancies. CHIP-harboring HSCs show a multilineage repopulation advantage over non-mutated HSCs [17, 18] and can exist as pre-leukemic stem cells (pre-LSCs) before the leukemic disease phenotype develops. Shlush et al. [18] proved that DNMT3A mutations occurred first, and IDH and NPM1 mutations occurred later in AML by showing that NPM1 and IDH mutations were detected in AML cells, whereas DNMT3A mutations were detected in non-leukemic cells (T lymphocytes) as well as myeloid cells of primary human AML samples.
Through these studies, the concept of AMLSC has differentiated into two developmental stages: pre-LSCs and LSC, which is also called leukemia-initiating cells. NGS studies have found that mutations in epigenetic/chromatin regulator genes, such as DTA (DNMT3A, TET2, and ASXL1), were substantially enriched in pre-LSCs. These mutations in epigenetic regulators may lead to aberrant transcriptional networks and the conversion of normal HSPC to pre-LSCs. These mutations presumably maintain these pre-LSCs in a “primed” state and can prepare for the way forvarious kinds of myeloid malignancies including AML. However, these pre-LSCs still contribute to normal multi-lineage hematopoiesis [19]. A “primed” pre-LSC is different from CHIP such that in an age-related clonal hematopoiesis, pre-LSCs have a propensity to transform into LSCs at the time of additional hits, while HSCs participating in CHIP do not. However, the mechanism that guides the different fates of these two similar features in pre-LSCs and CHIP-harboring HSCs with normal hematopoiesis is largely unknown. If additional hits are obtained in these pre-LSCs, they are converted to leukemia-initiating cells, which then become true “AMLSCs” by a traditional LSC concept. It is not until pre-LSCs develop into LSCs that they differentiate and proliferate aggressively into leukemic blasts and develop AML in the patient bone marrow (BM). For example, mutated DNMT3A-harbored HSCs transform into LSCs with the acquisition of the NPM1 mutation [20]. The current standard treatment strategy, involving administration of cytarabine arabinoside for 7 days and anthracyclines for 3 days, can effectively remove relatively differentiated leukemic cells with genetic mutations acquired sequentially during leukemogenesis but cannot eradicate pre-LSC clones. Epigenetic regulator gene mutations acquired during the pre-LSC development stage, such as DTA mutations, persist even after achieving morphologically complete remission after induction of chemotherapy [21, 22]. Therefore, the presence of DNMT3A and ASXL1 mutations is considered to be an adverse risk factor, which means that they are difficult to cure without allogeneic hematopoietic stem cell transplantation [23-25]. A staged leukemogenesis model of pre-LSC with a prior hit on an epigenetic regulator and LSC with a later hit on another gene in other signaling pathways is widely accepted and seems to be reasonable. However, it is not yet clear whether this leukemogenesis model is correct in most patients with AML in real world. Benard et al. [26] examined public databases with whole exome sequencing data and suggested a sequence of mutations based on the information of variant allele frequencies (VAFs) of each gene mutation. They suggested that a mutation of genes involved in chromatin modification, the cohesin complex, and DNA methylation preceded other gene mutations, including those involved in transcription and splicing, tumor suppressor genes, and NPM1 in many cases, while some cases showed that a precedent mutation in receptor tyrosine kinase/RAS signaling pathway genes or transcription related genes occurs first, followed by an additional mutation of genes involved in epigenetic regulators such as DNA methylation and chromatin regulation. These data are not direct evidence against the current model of pre-LSC and LSC, since they were obtained by a statistical algorithm with only VAF information of gene mutations. However, we can hypothesize that there may be at least multiple patterns of leukemogenesis in terms of the sequence of epigenetic and non-epigenetic gene mutations.
AMLSC IS NOT HOMOGENEOUS
CD34 expression is universally accepted as a basic marker for AMLSCs, and CD38 null or low expression of CD38 has been considered a reliable marker of AMLSCs. Many studies identifying putative LSC markers in AML have proposed candidate biomarkers for AMLSCs as those enriched in the CD34+CD38- cell fraction. T cell immunoglobulin mucin-3 (TIM-3) [27, 28], CD25, CD32 [29], CD96 [30], and C-type lectin-like molecule-1 (CLL-1) [31] have been suggested as LSC markers based on this old belief. However, in vivo functional validation of the LSC identification capabilities of these molecules is not always possible.
Ng and Dick performed an important study to investigate true LSC gene signatures. They sorted four fractions of primary AML cells using two surface markers, CD34 and CD38, and then split them for microarray and mouse transplant experiments. They analyzed the difference in gene expression between cells capable of repopulating NSG (NOD/SCID gamma) mice and cells incapable of repopulating in vivo [32]. In this study, AMLSCs were highly enriched in the CD34+CD38- cell population, but the other three fractions of CD34+CD38+, CD34-CD38+, and CD34-CD38- also contained LSCs, which means that the combination of CD34+ CD38- is not always a correct marker for AMLSCs. Flowcytometric analyses of primary AML samples demonstrate that AMLSCs with CD34+CD38- LSCs and CD34+CD38+ immuno-phenotypes are mixed in patients with AML [9, 33] and show remarkably heterogeneous features in terms of immunophenotype (Fig. 1).
Furthermore, several studies have suggested that LSCs reside within the CD34- cell population in certain AML cases. For example, some fractions of CD34- leukemic cells with PML-RARα fusion and NPM1 mutation have leukemia-initiating capacity [34, 35]. Certain mutations are associated with the CD34+CD38- dominant immunophenotype of multipotent progenitor (MPP)- or LMPP-like LSCs, whereas some mutations are associated with the CD34+CD38+ immunophenotype of GMP-like LSCs [33]. KRAS mutations were commonly observed in MPP-like LSC-dominant patients, whereas TP53 mutations were frequently observed in LMPP-like LSC-dominant patients. The observation that a certain genetic mutation is enriched in leukemic cells with a certain LSC-like phenotype suggests that each genetic mutation recurrently detected in AML might affect marker expression in AMLSCs. Alternatively, these mutations might initiate leukemia in terms of unlimited self-renewal capacity and immortality. For example, FLT3-ITD was enriched in CD34+CD38-CD123+ AML cells [36, 37], whereas PML-RARα transcript was enriched in CD34- cells [38]. As such, the genetics of AML cells is strongly linked to LSC phenotypes. AMLSCs are significantly heterogeneous in terms of immunophenotype and genomic context.
Recent advances in genomics have revealed an unexpectedly remarkable heterogeneity of genetic aberrations in AML. The Cancer Genome Atlas (TCGA) project opens mutational landscapes of AML, which shows a huge complexity in view of the numbers and combinations of recurrent genetic aberrations [14, 15]. According to these reports, the median number of mutated genes usually exceeds 3 in most AML cases, and the number of recurrently mutated genes is more than 30. As a result, the combination of these gene mutations causes remarkable heterogeneity in AML in view of the co-occurrence of gene mutations. When we consider combinations of triple gene mutations, the frequency of the most common combination of NPM1, DNMT3A and FLT-ITD was below 10% and that of the second most combination is below 5%, respectively [26]. A recent report using single-cell genomics technology clearly illustrated the striking diversity of leukemic clones in the same patient with AML, which further expanded our understanding of the heterogeneity of AMLSCs [39]. Thus, patients with AML having the exact same genetic profile, immunophenotypic profile, and clinical characteristics is rare.
HETEROGENEITY CHANGES OVER TIME
Current standard induction chemotherapy with cytarabine and idarubicin effectively induces complete morphological remission in approximately 70% of patients with newly diagnosed AML [40]. However, a significant proportion of patients treated with intensive chemotherapy and subsequent allogeneic hematopoietic stem cell transplantation experience relapse or refractoriness. A treatment strategy with chemotherapy regimens similar to that in the initial treatment is usually ineffective because leukemic clones resistant to the initial treatment expand through clonal evolution or surviving leukemic clones gain new mutations [41]. Shlush et al. [18] studied gene mutation occurrence in hematopoietic hierarchical cell clusters at the initial diagnosis and at the time of relapse. They found two patterns of relapse, termed “relapse origin-primitive” (ROp) and “relapse origin-committed” (ROc). In ROp, a small pool of LSCs generates bulk blasts, and the seeds of relapse are buried within these rare LSCs. At relapse, these LSCs proliferate and differentiate into blasts with a primitive phenotype with increased stemness programs, which reflects a switch towards a shallower developmental hierarchy upon leukemic progression. In contrast, in ROc, relapse originates from LSCs in larger subclones that have a committed immunophenotype and retain strong stem-cell-like or stemness transcriptional programs.
Another study revealed that LSC frequencies are remarkably increased (up to 90 folds) at the time of relapse in AML. However, the LSC marker frequencies do not change accordingly [42]. In this study, among the various markers, including CD32, CD33, CD45RA, CD47, CD96, CD97, CD99, CD123, HLA-DR, interleukin-1 receptor accessory protein (IL-1RAP), and TIM-3, only TIM-3 and CD96 showed increasing tendencies without statistical significance. TIM-3 expression levels were increased in patients with AML with failed chemotherapy [42]. In addition, there is evidence of plasticity of LSC surface markers in the same patient over time. For example, CD25+ LSC were shown to give rise to progeny of CD25- LSC capable of leukemic engraftment in serial transplantation assays in patient-derived xenograft models [43].
In summary, AMLSCs can change during treatment through clonal selection, plasticity of surface markers, and modification of the stemness transcriptional programs, even without increased LSC marker expression.
AS LONG AS RESEARCHERS PURSUE A UNIVERSAL TARGET, THERE IS A HOPE
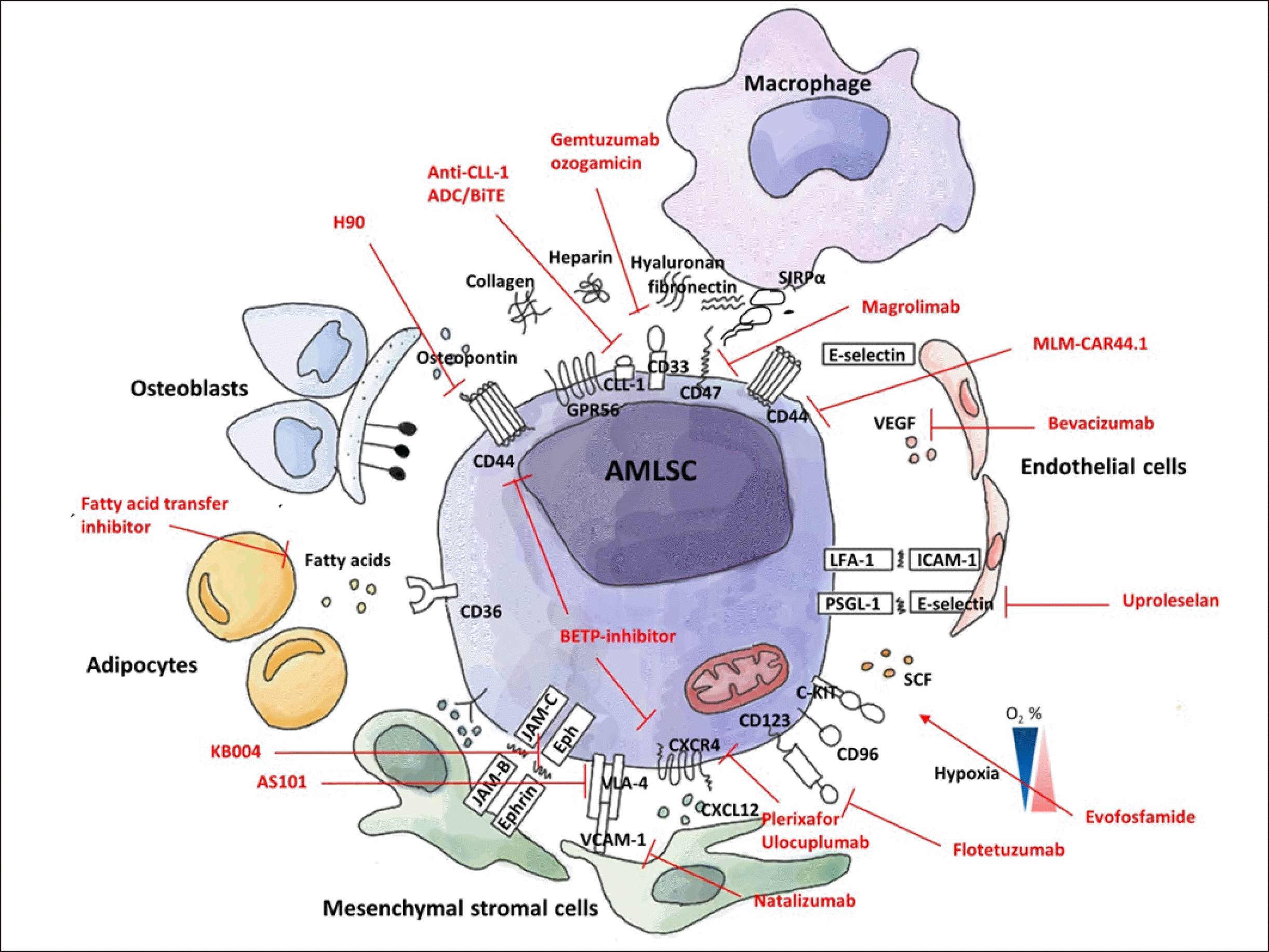
Although there has been a lot of evidence of heterogeneity from various standpoints of immunophenotype, cell cycle, metabolism, and stemness, there is still hope for an effective targeting strategy of a universal AMLSC biomarker. The Dick JE group identified 17 significant AMLSC genes through gene expression profiling selected based on results from microarray analysis and engrafting capability by xenotrans-plantation assay using samples from 78 patients with AML [32]. 17-LSC signature genes also include genes expressed in normal HSCs, such as CD34 and GPR56. The 17-gene LSC score is also well correlated with the European LeukemiaNet risk classification widely used for the risk stratification of patients with AML in current clinical practice [44]. Jung and Majeti investigated the methylated gene profiles in LSCs capable of being engrafted into NSG mice compared to those in leukemic blast cells [45] because stemness in HSCs and LSCs is known to be regulated by epigenetic mechanisms [46]. They found that AMLSCs are hypomethylated compared to blasts, and the AMLSC epigenetic signature is largely mutation-independent and uniquely characterized by hypomethylated and upregulated HOXA cluster genes HOXA9, HOXA7, and HOXA10 [45]. Based on the belief that there must be a universal LSC-unique biomarker for AML, continuous efforts to identify an AMLSC biomarker have been made worldwide. CD45RA was once reported as a prognostic LSC marker [47]. CD96 and CD99 are also promising markers because they are enriched in CD34+ CD38- AML blasts that show engraftment capacity and are not frequently expressed in normal HSCs [30, 48]. CD123, the interleukin-3 receptor alpha chain, has been proposed as an LSC marker to distinguish LSCs from HSCs [49]. Flotetuzumab (anti-CD123/CD3, previously known as MGD006), a humanized dual affinity re-targeting molecule, showed promising efficacy, especially in patients with TP53-mutated AML [50]. CLL-1, a transmembrane glycoprotein, one of the candidate AMLSCs, can be an effective target for LSC-directed therapy because CLL-1 is enriched in AML primary samples, whereas it is absent in uncommitted CD34+CD38- or CD34+CD33- normal stem and progenitor cells [51]. TIM-3 is another marker to differentiate LSCs from HSCs [28]. Targeting TIM-3 impairs LSC function but not HSC engraftment [27]. CD47, a critical innate macrophage immune checkpoint known to be a “do not eat me” signal, binds to signal-regulatory protein α (SIRPα) and prevents phagocytosis by macrophages. CD47-directed therapy using monoclonal antibodies has shown promising efficacy [52] and is currently being investigated in a phase 3 clinical trial. Some of these markers, such as CLL-1 and IL-1RAP, are thought to be AMLSC-specific, whereas some markers, including CD44, CD47, Roundabout4 (ROBO4), C-X-C motif chemokine receptor 4 (CXCR4), and CD33, are shared among normal HSPCs [53].
LSC NICHE PROTECTS AMLSCS FROM CHEMOTHERAPY
The stemness program that is strongly expressed in AMLSCs is determined by both cell-intrinsic and cell-extrinsic mechanisms. Cell-extrinsic factors are derived from the microenvironment or niche in which the cell resides. LSCs share cellular niche components with healthy HSCs in the BM, including osteoblasts, endothelial cells, mesenchymal stromal cells (MSCs), adipocytes, and sympathetic neurons. Several factors such as stem cell factor (SCF), C-X-C motif chemokine ligand 12 (CXCL12), Notch ligands, and transforming growth factor-β are produced by these niche cells and are involved in the maintenance of HSCs [54]. AML cells not only share a niche with HSCs for their survival but also affect HSC niche conditions in various ways. Adipocytes abundant in normal BM are important cell components of the HSC niche; however, their role is disrupted in an AML xenograft model [55], MSCs are also important for HSC maintenance through the secretion of various factors such as SCF and CXCL12. In an MLL-AF9 mouse model, the profile of MSCs is changed to an osteoblastic one, and the production of SCF and CXCL12 is decreased. Altered microenvironments in turn provide a competitive growth advantage for LSCs over HSCs. Thus, a friendly niche acts as an enemy’s shield to protect LSCs from chemotherapy-induced apoptosis by maintaining a quiescent status [56]. This “leukemia-permissive” microenvironment is one of the critical causes of therapy resistance and a cause of suppressed normal hematopoiesis in AML.
Leukemic cells attach to the stromal ligands vascular cell adhesion molecule (VCAM-1), fibronectin, and intracellular adhesion molecule 1 (ICAM-1) of the niche via the interaction of the β-1 integrin receptor family members very late antigen-4 (VLA-4) and VLA-5, and the β-2 integrin LFA-1 [57]. The homing receptor CXCR4 guides LSCs to CXCL12 in the BM niche. The therapeutic targeting of the CXCL12-CXCR4 interaction has shown efficacy as an adjunctive therapy to eradicate AMLSCs [58]. Bromodomain and extra-terminal domain-containing (BET-containing) protein (BETPs) inhibitors degrade BETPs, downregulate CXCR4 and CD44 expression, and decrease the LSC population in a patient-derived xenotransplantation model [59]. CD44, an extensively alternatively spliced adhesion molecule, mediates adhesive cell-cell and cell-extracellular matrix interactions by binding to its ligand hyaluronan in the endosteal region or osteopontin, fibronectin, and selectin. Targeting CD44 by monoclonal antibody showed promising results in effectively targeting LSCs and reversing the AML differentiation block [60].
Hypoxia is a well-known microenvironmental factor in HSCs/LSCs [61]. Mantel and Broxmeyer et al. [62] demonstrated that exposure to extraphysiologic oxygen stress reduced the yield of HSCs harvested from BM by a series of delicate experiments including BM HSC harvest and functional assays under strictly controlled hypoxic conditions. A major regulator of hypoxia, HIF-1α signaling promotes the expression of VEGF, CXCR4, CXCL12, and SCF in both AML blasts and stromal cells [63]. The synthesis of HIF-1α in the hypoxic BM microenvironment induces upregulation of CXCR4 expressed on LSCs, thereby contributing to the protection of LSCs in the BM niche by their anchorage. In patients with AML with mutated IDH1/2, the accumulated oncometabolite 2-hydroxyglutarate (2-HG) results in an increased reactive oxygen species generation and activation of HIF-1α [64]. Another regulator of hypoxia, HIF-2α, also plays an important role in the survival of primary AML cells. HIF-2α-deficiency displays decreased engraftment ability of human AML cells and accelerates AML progression [65, 66]. Hypoxia-activated prodrugs designed to be activated in the hypoxic LSC niche are expected to be promising; however, they are not yet successful [67].
The BM contains a large pool of adipocytes, and leukemic cells use fatty acids as important fuels. Therefore, adipocyte targeting is being actively investigated. Restoring normal adipocyte maturation using PPARγ agonists and inhibiting fatty acid transfer into cells has been explored as future therapies [68].
In addition to the niche factors described above, many adhesion molecules and related pathways, including integrins/CD98, CD44/E-selectin, Eph, cadherin, GPR56, and JAM-C, are being investigated as possible future LSC niche-directed therapies (Fig. 2).
CONCLUSION
AMLSCs originating from HSPCs are the source of a more differentiated leukemic bulk and the cause of relapse and refractoriness despite the current standard treatment, which includes intensive chemotherapy and allogeneic hematopoietic cell transplantation. The discovery of the CD34+ CD38- immunophenotype as an LSC marker introduced the first model of AMLSC as a conceptual pathologic counterpart of normal HSPC. Studies in mice have shown that AMLSCs originate from hematopoietic stem cells to progenitor cells. Methodologies of multicolor flow cytometry and NGS have made deep investigations of small volumes of primary AML samples possible, which introduced the concept of pre-LSCs prior to the development of leukemia-initiating stem cells. However, the current concept is not perfect, and huge heterogeneities in terms of mutational and immunophenotypic profiles among and within patients have been revealed. Genetic aberrations detected in AML at the initial diagnosis change at the time of relapse through clonal evolution by selective survival pressure induced by cytotoxic chemotherapy. AMLSC frequencies increase during treatment over time, and the stemness program of AMLSCs becomes shallower, making patients with relapsed AML more therapy-resistant. AMLSCs have relatively common epigenetic and genetic signatures despite their huge heterogeneity. Many putative AMLSC markers have been proposed, and efforts to target AMLSCs have been made. Recent approaches to target AMLSCs and their niche have shed light on promising future treatment strategies to cure AML.

 XML Download
XML Download