

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Thrombotic microangiopathy (TMA) is a potentially fatal syndrome characterized by microangiopathic hemolytic anemia (MAHA), consumptive thrombocytopenia, and variable ischemic end-organ injury. TMA is a heterogeneous group of diseases, including thrombotic thrombocytopenic purpura (TTP), atypical hemolytic uremic syndrome, and secondary TMA caused by abnormal coagulation, infection, malignancy, transplantation, drug, autoimmune disease, or pregnancy (Table 1) [1, 2].
TTP is defined as a severe deficiency (activity <10%) of ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13). ADAMTS13 is a plasma metalloprotease that cleaves von Willebrand factor (vWF), which mediates platelet adhesion to damaged vascular endothelium and platelet aggregation. ADAMTS13 deficiency leads to the accumulation of unusually large vWF multimers in the bloodstream and subsequent exacerbation of platelet adhesion, aggregation, and formation of microthrombi in small arterioles and capillaries [3-7]. TTP is divided into congenital (inherited) and immune-mediated (acquired) TTP. Congenital TTP (also called Upshaw–Schulman syndrome) is caused by biallelic mutations in the ADAMTS13 gene [8]. Immune-mediated TTP (iTTP) is an autoimmune disorder resulting from the production of autoantibodies against ADAMTS13 [1-3]. TTP is a rare disease with an annual incidence of approximately 2–6 cases/million/year [9, 10]. TTP typically presents in adulthood, with a median age at onset around the fourth decade of life. iTTP is 2 to 3 times more frequent in women [3]. Congenital TTP accounts for approximately 3–5% of all cases of TTP [9].
A TTP-like syndrome can be observed in patients with critical conditions, which cause vascular endothelial injury, such as sepsis, trauma, and cancer. Damage to the vascular endothelium leads to the enhanced release of unusually large vWF. Excessive unusually large vWF become anchored to endothelial cells of targeted organs and recruit activated platelets to assemble unusually large vWF-platelet complexes. Both TTP-like syndrome and TTP produce similar pathological conditions of microvascular thrombosis. However, the former involves normal hemostasis in endothelial injury and the latter involves pathologic hemostasis without endothelial injury [11].
Recently, remarkable progress has been made in understanding the pathophysiology of iTTP. This has been successfully translated to patient management. This study aimed to review recent advances in the diagnosis, treatment, and long-term follow-up of patients with iTTP.
DIAGNOSIS AND CLINICAL PREDICTION MODELS
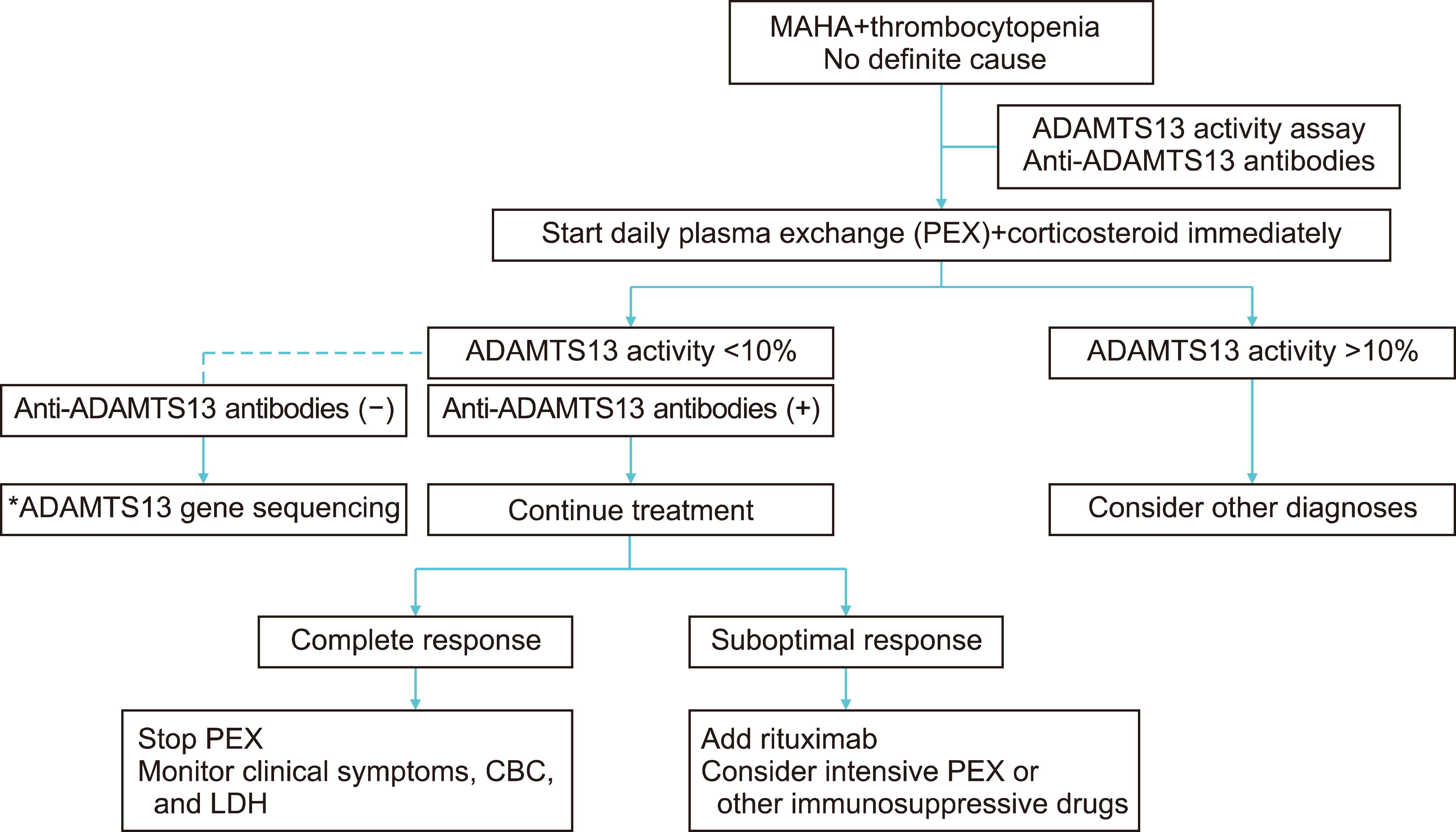
ADAMTS13 activity <10% confirms the diagnosis of TTP. ADAMTS13 activity should be tested in all patients with MAHA and thrombocytopenia without a definite cause. A blood sample for the ADAMTS13 assay must be collected before the initiation of treatment to avoid the false-negative result. ADAMTS13 assay is not immediately available in most centers in Korea, and several days are required to obtain the results. TTP is a medical emergency, and a delay in appropriate therapy is associated with substantial morbidity and mortality. Immediate initiation of treatment may be lifesaving before the ADAMTS13 activity level is known. Patients with TTP uniformly present with severe thrombocytopenia (<30×109/L) and MAHA with evidence of schistocytes on a peripheral blood smear. Parameters of hemolysis are also present, including elevated reticulocyte count, elevated lactate dehydrogenase (LDH) level, elevated unconjugated bilirubin level, and undetectable haptoglobin concentration. Coombs’ test is usually negative, and coagulation parameters are usually normal [3].
Early recognition of TTP is difficult because of its diverse clinical manifestations. Approximately 60% of patients with TTP have neurological symptoms that range from headache to stroke, seizures, or coma [9, 12-16]. Mesenchymal ischemia is present in 35% of patients [9]. Approximately 25% of patients have cardiac symptoms, ranging from electrocardiographic abnormalities to myocardial infarction [17]. Although acute renal injury is common in TTP, severe renal failure is rare in iTTP [18]. The Korean TTP registry reported symptoms, such as fever (56%), bleeding (45%), and severe neurological symptoms (20%), upon admission [16].
Clinical prediction of TTP is important to make initial management decisions before ascertaining the ADAMTS13 activity level. The French and PLASMIC scores have been developed and validated to estimate the probability of severe ADAMTS13 deficiency (Table 2) [19, 20]. These prediction scores incorporate basic clinical and laboratory parameters. Both scores showed that severe thrombocytopenia and mild renal impairment were strongly associated with a severe ADAMTS13 deficiency. The PLASMIC score includes not only platelet count and creatinine levels, but also the evidence of hemolysis, presence of active cancer, history of transplantation, mean corpuscular volume, and international normalized ratio. When the PLASMIC score was applied to the external validation cohort, 82% of patients with a PLASMIC score of 6 or 7 had severe ADAMTS13 deficiency compared with 4% of patients with a score of 0–4 [20]. A PLASMIC score ≥5 suggests the requirement of immediate treatment, whereas a low probability score requires an alternative diagnosis. However, it is important to recognize that clinical prediction scores are accurate only when applied to a population with TMA without an obvious cause, and these models cannot definitively confirm TTP.
Detection of anti-ADAMTS13 antibodies confirms the diagnosis of iTTP. However, a negative anti-ADAMTS13 antibody assay does not exclude iTTP because antibodies become detectable during the follow-up period or relapse. In cases that have persistent severe ADAMTS13 deficiency with no detectable anti-ADAMTS13 antibodies during remission, ADAMTS13 gene sequencing should be performed to rule out congenital TTP [3].
TREATMENT OF ITTP
Current treatment in Korea: plasma exchange and corticosteroids
Before the use of plasma exchange (PEX), the survival rate of patients with iTTP was 10–20%. Therapeutic PEX has improved survival rates from <20% to >80%. The Canadian Apheresis Group conducted a prospective randomized study, comparing PEX with plasma infusion for acute TTP treatment. They reported that patients receiving PEX had a higher response rate, lower early mortality rate (3.9%), and improved survival at 6 months (78%) [21]. The effectiveness of PEX is related to the supply of ADAMTS13 with removal of unusually large vWF multimers and anti-ADAMTS13 autoantibodies. Given the high risk of early death in TTP, PEX treatment should be initiated at the earliest early. The optimal PEX regimen has not yet been determined. However, according to the Canadian apheresis trial group and guidelines from the British Committee for Standards in Haematology, PEX is usually started by a 1.5-plasma volume exchange for the first 3 days, followed by a 1.0 plasma volume exchange [12, 22]. Daily PEX should be continued for a minimum duration of 2 days after platelet count normalization. More intensive exchanges, such as twice-daily PEX, should be considered in resistant disease.
Corticosteroids suppress anti-ADAMTS-13 autoantibodies [23]. In addition to PEX, corticosteroids should be administered to all patients with an acute episode of iTTP. Although the benefits of corticosteroids were not well determined in a randomized clinical trial, the use of corticosteroids may be supported by the autoimmune nature of the disease. Corticosteroids have been administered as either intravenous pulse methylprednisolone (1 g/day for 3 days, followed by 1 mg/kg/day prednisone) or oral prednisolone (1 mg/kg/day) (Fig. 1) [22].
The criteria for clinical response, exacerbation, remission, and relapse of TTP have been defined by the International Working Group (IWG) for TTP in 2017 [2]. ‘Clinical response’ was defined as a sustained platelet count increment (platelet count >150×109/L) and LDH <1.5 times the upper limit of normal (ULN). ‘Refractory iTTP’ is defined as a lack of platelet count increment (platelet count <50×109/L) with persistently high LDH levels (>1.5×ULN) despite undergoing PEX five times or a decrease in platelet count after an initial improvement while receiving PEX. ‘Exacerbation’ refers to early recurrence of iTTP within 30 days of discontinuing PEX. ‘Clinical remission’ of iTTP is defined as a sustained clinical response for more than 30 days after discontinuation of PEX. ‘Relapse’ is defined as a decrease in platelet count to below the lower limit with or without clinical symptoms, after clinical remission [2]. Relapses occur in 30–50% of patients with iTTP [3]. Despite advances in iTTP management, refractory disease and recurrence remain critical issues.
Rituximab
Rituximab, a monoclonal antibody targeting CD20 on B cells, has been used to treat iTTP by suppression of ADAMTS13 autoantibody production. Several groups have conducted studies to demonstrate the efficacy of rituximab for refractory or recurrent iTTP [24-28]. A UK group reported that clinical remission was achieved in all 25 patients who received rituximab, and relapses were not observed during a median follow-up of 19 months [24]. In a prospective study involving 40 patients with refractory (N=20) or relapsed iTTP (N=20), rituximab treatment resulted in remission in 63.6% of refractory patients and 90% of relapsed patients at 8 weeks [26]. The Korean Society of Hematology Thrombosis Working Party conducted a nationwide survey to validate whether rituximab improved outcomes in Korean patients with iTTP. Rituximab successfully induced clinical remission in 10 of 12 patients (83%) who were refractory to PEX. During a median follow-up of 79 weeks, there was no relapse in 10 responders [28]. Although rituximab therapy for iTTP has not yet been approved by the Korean Ministry of Food and Drug Safety (MFDS), rituximab should be considered for refractory or relapsed patients with iTTP for “off-label use” after obtaining approval from each Institutional Review Board and the MFDS.
Front-line rituximab in conjunction with standard treatment for acute iTTP significantly reduced the relapse rate (10% compared with 57% in historical controls) [29]. Earlier upfront administration of rituximab within 3 days may reduce the time to remission, number of PEX days, and days of hospitalization [30]. Rituximab can be used as a preemptive therapy to prevent relapse after the detection of ADAMTS13 deficiency during follow-up [31, 32]. The development of severe ADAMTS13 deficiency during remission is significantly associated with a high risk of relapse [32-35]. A French group reported the efficacy of preemptive rituximab for iTTP patients with lower ADAMTS13 activity (<10%) during remission. There was a significant reduction in the relapse rate in the preemptive treatment cohort, compared with that in historical controls [32].
Measurements of ADAMTS-13 at regular intervals during treatment and remission (e.g., weekly during treatment, monthly, and then every 3 months during the follow-up period, extending to every 6–12 months) may provide data about the risk of relapse and persistence of subclinical disease activity [2].
Other immunosuppressive therapies
Other immunomodulators, such as vincristine, cyclosporine, and cyclophosphamide, have been used as second-line therapies for patients with refractory iTTP, pre-rituximab era [36]. The clinical response rate was 50–87% in patients who received vincristine. Additionally, bortezomib, a proteasome inhibitor, was found to be effective in the treatment of refractory or relapsed iTTP by depleting plasma cells [36, 37]. Among 12 patients with iTTP who received bortezomib as a salvage treatment, 11 survived and maintained remission [37].
Caplacizumab
Caplacizumab, an anti–vWF humanized single-variable domain immunoglobulin (nanobody), targets the A1 domain of vWF, inhibits vWF-mediated platelet aggregation, and subsequently prevents microvascular thrombosis [38]. The efficacy and safety of caplacizumab in patients with iTTP have been evaluated in the phase 2 TITAN and phase 3 HERCULES trials [39, 40]. The median time to normalization of platelet count was shorter with caplacizumab than with a placebo (2.69 days vs. 2.88 days, P=0.01) in the HERCULES trial. The caplacizumab group required less PEX and had a shorter hospital stay than the placebo group [40]. The percentage of patients with a composite outcome of TTP-related death, recurrence of TTP, or a major thromboembolic event was lower with caplacizumab than with a placebo (12% vs. 49%, P<0.001) [40]. In the integrated analysis of data from both trials, a significant reduction in the number of deaths (0 vs. 4; P<0.05) and a significantly lower incidence of refractory TTP (0 vs. 8; P<0.05) were observed in patients who received caplacizumab [41]. The most common adverse event associated with caplacizumab was mucocutaneous bleeding. However, these events were mild or moderate in severity, and resolved spontaneously in most patients [39-41].
Caplacizumab (10 mg intravenous loading bolus, followed by 10 mg daily subcutaneously) was administered for 30 days after the last daily PEX in the phase 2 TITAN trial. The relapse rate after the discontinuation of caplacizumab was 22% in this trial. A subgroup of patients with persistent severe ADAMTS13 deficiency (<10%) after discontinuation of caplacizumab had a higher risk of relapse [39]. Based on these results, the HERCULES trial was designed to monitor ADAMTS13 weekly and use caplacizumab for patients with ADAMTS13 deficiency (<10%) for up to 4 weeks at the end of the treatment period. The relapse rate after discontinuation of caplacizumab was 8% in the HERCULES trial. In patients who receive caplacizumab, monitoring of ADAMTS13 activity may be used to tailor the duration of therapy. Although the threshold for discontinuation of therapy has not been well defined, ADAMTS13 >20% for at least 2 consecutive weeks may be a reasonable point [42].
Recently, the IWG proposed revised consensus outcome definitions that incorporate ADAMTS13 activity and the effects of anti-VWF therapy [43]. In this proposal, clinical remission is defined as a sustained clinical response with either no PEX and no anti-vWF therapy for ≥30 days or with the attainment of ADAMTS13 remission (partial or complete). Partial ADAMTS13 remission is defined as ADAMTS13 activity ≥20% to less than the lower limit of normal (LLN). Complete ADAMTS13 remission refers to ADAMTS13 activity ≥LLN. ADAMTS13 relapse is termed when ADAMTS13 levels decrease to <20% after ADAMTS13 remission [43].
Novel agents under clinical trials
Recombinant human ADAMTS13 (rhADAMTS13) has been shown to normalize the cleavage of vWF from congenital TTP plasma [44]. The safety and pharmacokinetics of rhADAMTS13 have been investigated in patients with severe congenital TTP. In a phase I trial, rhADAMTS13 was safe, non-immunogenic, and well tolerated. The pharmacokinetic profile was comparable to that of plasma infusion [45]. This drug has been granted a fast-track designation by the US Food and Drug Administration for the treatment of congenital TTP. In vitro addition of rhADAMTS13 to plasma can override the inhibitor and cleave vWF in iTTP [44]. A clinical trial (NCT03922308) is ongoing to evaluate the efficacy of rhADAMTS13 in iTTP.
Anfibatide, a snake C-type lectin derived from the venom of Agkistrodon acutus, binds to the platelet receptor GPIb and prevents its adhesion to vWF. Anfibatide inhibited the interaction between vWF multimers and platelets without a significant increase in bleeding tendency in healthy controls [46]. A randomized phase II study (NCT04021173) is ongoing to evaluate the safety and efficacy of anfibatide for iTTP.
LONG-TERM OUTCOMES AFTER RECOVERY FROM AN ACUTE EPISODE OF ITTP
With prompt and effective treatment, most patients with iTTP survive an acute episode. Although the major concern following recovery from an acute episode of iTTP is the risk of relapse, a higher rate of chronic morbidities, poor quality of life, and a higher mortality rate are problems in TTP survivors [47-51]. Patients enrolled in the Oklahoma TTP-HUS Registry had an increased prevalence of hypertension, cognitive impairment, and major depression compared with the reference populations. These patients also had a higher risk of premature death [47]. Several groups have also shown TTP survivors have a higher incidence of other health complications, including major adverse cardiovascular events, neurocognitive deficits, and autoimmune disorders than the general population [48-51]. These chronic morbidities may have a significant impact on quality of life. Moreover, TTP survivors without significant disabilities or additional health problems consistently scored lower across all domains of health-related quality of life surveys than the general population [48, 49]. Cardiovascular disease is the leading cause of death among iTTP survivors. Upreti et al. [50] demonstrated that patients with iTTP had a five-fold higher stroke prevalence than age- and sex-matched controls. Another study reported that the prevalence of major adverse cardiovascular events during iTTP remission was 28.6% at a median follow-up of 7.6 years, with the first major adverse cardiovascular events occurring 1–2 decades earlier than in the general population [51]. Aggressive screening and management of cardiovascular risk factors may reduce the incidence of cardiovascular events in iTTP survivors. Meticulous long-term follow-up of iTTP survivors is crucial in identifying the development of additional health problems.
CONCLUSIONS
PEX and corticosteroids, the mainstay of treatment for acute episodes of iTTP in Korea, have dramatically improved the survival of patients with iTTP. However, mortality rates have been reported to be 10–20%. A subset of patients remains refractory to PEX, and relapses after discontinuation of PEX. Prompt initiation of treatment is crucial for reducing early death. Targeted therapies based on iTTP pathogenesis will help overcome these unmet needs. Rituximab has shown to be effective for the treatment of refractory or relapsed iTTP, and may be useful as a first-line or pre-emptive therapy. Caplacizumab shortened the time to normalization of platelet count and reduced refractoriness, recurrence, and death during the acute phase of iTTP. ADAMTS13 activity is an important predictor of recurrence and can guide the duration of anti-vWF therapy. iTTP survivors are at risk of several long-term complications, including cardiovascular disease, depression, and neurocognitive deficits, which can lead to poor quality of life and shorter life expectancy. We need to investigate the mechanisms, risk factors, and management of these complications to improve the long-term outcomes of iTTP.
 XML Download
XML Download