

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
MGRS
MGRS-RELATED KIDNEY DISORDERS WITH MONOCLONAL IMMUNOGLOBULIN DEPOSITS
Lesions with organized deposits
Lesions with non-organized deposits
MGRS-RELATED KIDNEY DISORDERS WITHOUT MONOCLONAL IMMUNOGLOBULIN DEPOSITS
NEUROLOGIC MGCS
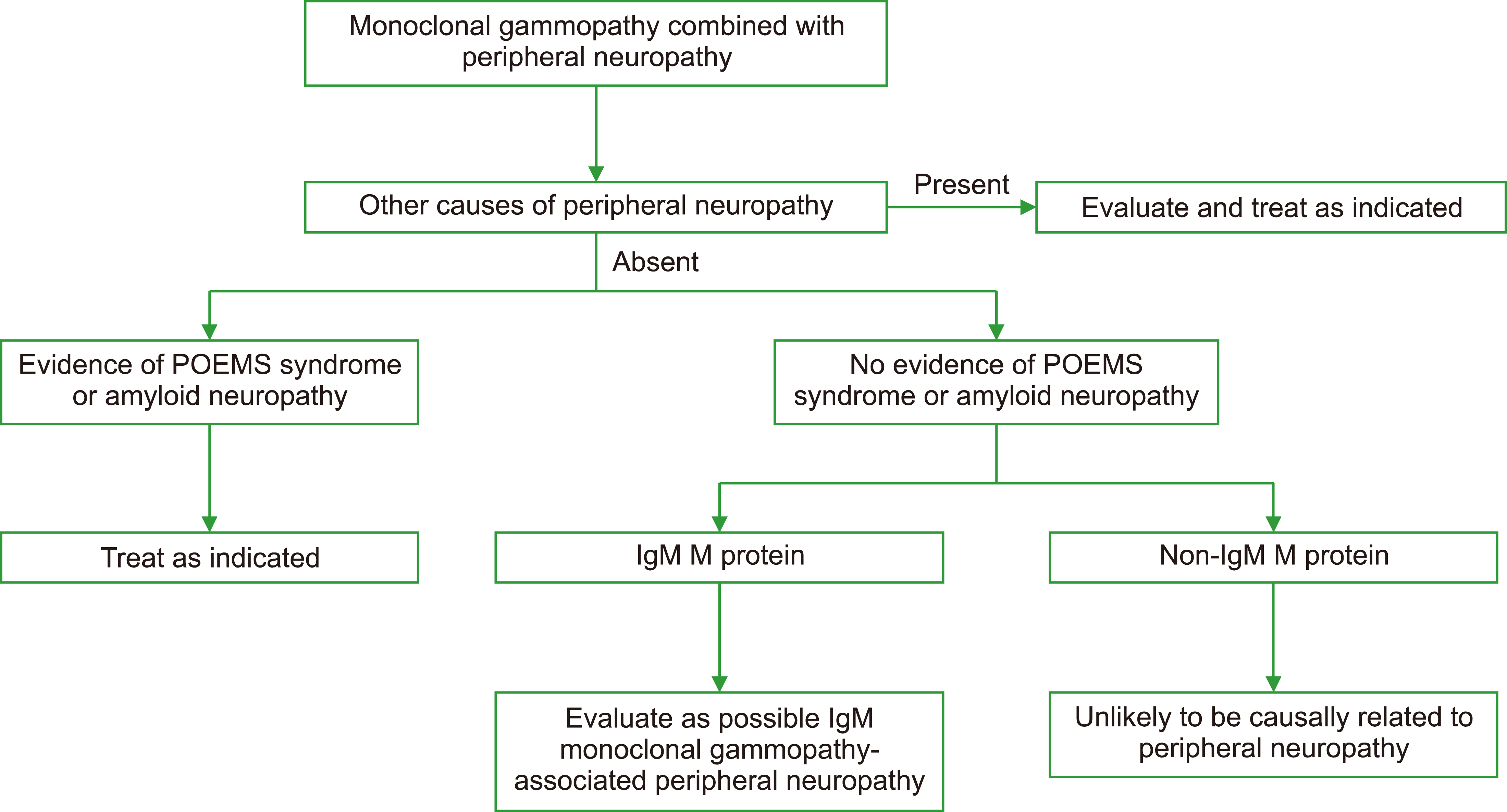
CUTANEOUS MGCS
Table 1
| POEMS syndromea) [26] | Scleromyxedema [24] | Schnitzler syndromeb,c) [26] | Necrobiotic xanthogranulomad) [30] | TEMPI syndrome [33] |
|---|---|---|---|---|
|
Mandatory major criteria |
Obligate criteria |
Major criteria |
Major criteria |
|
|
Other major criteria |
||||
Minor criteria
|
Minor criteria |
Minor criteria |
Minor criteria |
a)POEMS syndrome diagnosis is confirmed when both mandatory major criteria, one of the other major criteria, and one of the minor criteria are present. b)Definite diagnosis of Schnitzler syndrome: if IgM, both obligate criteria and at least2 minor criteria; if IgG, both obligate criteria and 3 minor criteria. c)Probable diagnosis of Schnitzler syndrome: if IgM, both obligate criteria and 1 minor criteria; if IgG, both obligate criteria and 2 minor criteria. d)Xanthogranuloma diagnosis is confirmed when both major criteria and at least 1 minor criterion are present. It is only applicable in the absence of a foreign body, infection, or other identifiable causes.
![]()
 XML Download
XML Download