

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Inherited platelet disorders (IPDs) can cause mucocutaneous bleeding due to impaired primary hemostatic function of platelets, thrombocytopenia, or both. IPDs show markedly heterogeneous clinical presentation and genetics [1-3]. The prevalence of IPDs is considered very low, approximately 1 in 104-6 persons worldwide [4, 5]. In Korea, the exact prevalence is unknown, although sporadic cases have been reported [6, 7].
Generally, IPDs can be classified based on their altered platelet function. However, in many instances, a definite distinction between disorders of platelet adhesion, activation, secretion, aggregation, and procoagulant activity is doubtful [6, 8]. Therefore, the classification of IPDs according to abnormalities of platelet components that share common features has been suggested (Table 1) [8]. Although this classification can help understand the pathophysiology and characteristics of each IPD, a specific subtype of IPD is diagnosed by synthesizing various test results in real-world clinical practice. A few clinical guidelines suggest a step-by-step approach for diagnosing IPDs [2, 3, 9, 10]. The first step in the diagnosis of IPDs starts with a careful clinical evaluation of the patient and family members, including history and pattern of bleeding with or without surgery and trauma, intake of food drugs that can affect platelet function, and family history of thrombocytopenia or other malignancies [9, 11]. Since bleeding symptoms can be subjective in both patient descriptions and physician examinations, bleeding assessment tools (BAT) can standardize the severity of bleeding symptoms [9, 11, 12]. In the clinical setting of von Willebrand disease (vWD), an international society of thrombosis and hemostasis (ISTH) BAT score >6 showed a 99% probability of an IPD [11, 13]. In addition, a complete physical examination should be performed to evaluate signs of bleeding and the features of potential syndromic IPDs [9]. If IPDs are suspected, a laboratory workup begins. Routine laboratory evaluations, such as complete blood count (CBC), prothrombin time, activated partial thromboplastin time, and screening tests for vWD including von Willebrand factor (vWF) antigen, ristocetin cofactor activity, and coagulation factor VIII activity, should be performed [2, 9]. Diseases that are not related to platelet function, particularly immune thrombocytopenia (ITP), should be excluded before starting diagnostic workup for IPDs. In ITP, severe and spontaneous bleeding usually occurs when the platelet count falls less than 20,000/µL [11, 14]. If the preliminary laboratory results show no abnormalities, a diagnostic workup for IPDs should be initiated. In this review, currently available laboratory tools for diagnosing IPDs and a diagnostic algorithm based on this information are presented.
LABORATORY TOOLS FOR THE DIAGNOSIS OF IPDS
Complete blood count (CBC) and peripheral blood (PB) smear morphology
CBC and PB smears should be performed as the first-step tests. The number of platelets provides important information to guide further workup processes for diagnosing IPDs. If there is a normal platelet count, but IPDs are suspected, evaluation of platelet function is recommended [4]. If the patient shows thrombocytopenia, platelet size should guide further evaluation [4]. Microthrombocytopenia (thrombocytopenia with small platelet size) includes Wiskott-Aldrich syndrome (WAS) and x-linked thrombocytopenia (XLT) [4, 8]. Thrombocytopenia with normal platelet size is related with various IPDSs and acquired disorders, and macrothrombocytopenia (thrombocytopenia with large platelet size) can be seen in Bernard-Soulier syndrome (BSS), MYH9-related disease, 22q deletion syndrome, and Paris/Trousseau/Jacobsen syndrome (PT/J) [4, 8, 11]. Platelet size can be estimated using the mean platelet volume (MPV), and the presence of giant platelets is usually flagged by automated hematologic analyzers [15]. According to a study, an MPV >12.4 fL showed 83% sensitivity and 89% specificity to differentiate IPDs and ITP [11, 16]. In addition, recent automated hematological analyzers provide another platelet index, the immature platelet fraction (IPF), relative to mature platelets [17]. Immature platelets are the most recently produced platelets released into PB from the bone marrow and are an analog of reticulocytes in terms of platelets [18]. Since the number and proportion of immature platelets reflect the rate of thrombopoiesis, they represent thrombopoietic activity [18]. IPF can distinguish IPDs from thrombocytopenia due to acquired marrow failure [11]. A study reported that IPF tends to be higher in IPDS than in immune thrombocytopenia and other causes of thrombocytopenia; however, the diagnostic sensitivity and specificity should be further evaluated [11, 19].
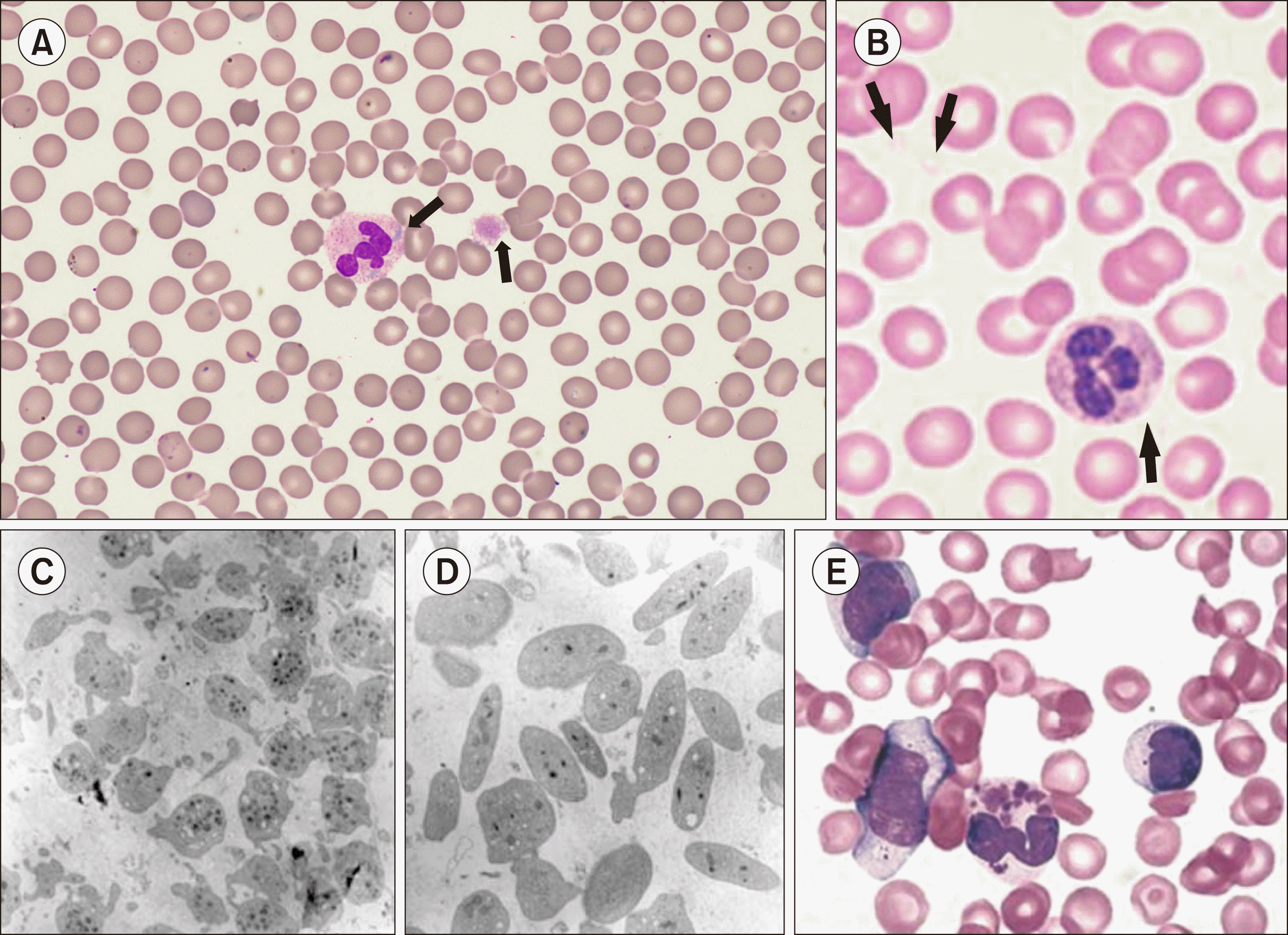
Information regarding platelet count, size, and morphology can be obtained by PB smears. Although CBC can provide information on platelet count and size, it may be inaccurate in the setting of macrothrombocytopenia and microthrombocytopenia [4]. Platelet size has diagnostic significance when considered in relation to platelet count [15]. It can be roughly calculated by comparing the diameter with normal red blood cells. Thrombocytopenia with the presence of large (3–7 mm in diameter, roughly the diameter of a normal-sized red blood cell) or giant (10–20 mm in diameter, larger than normal-sized red blood cell) platelets suggests macrothrombocytopenic IPDs (Fig. 1A) [15]. ‘Gray’ or ‘pale’ platelets, which mean decreased or absence of platelet granules, can be an important clue of gray platelet syndrome (GPS) (Fig. 1B–D). Additionally, the morphology of other blood cells can provide valuable information for further evaluation. For example, Döhle-like inclusion bodies in neutrophils are a characteristic finding of MYH9-related disease (Fig. 1A), and abnormal RBC morphology suggests a GATA-1 mutation [4]. In addition, giant neutrophilic granules suggested the presence of Chediak-Higashi syndrome (CHS) (Fig. 1E) [8, 11].
Platelet function tests
Light transmission aggregometry (LTA) is the most widely used platelet function test [11]. LTA uses platelet-rich plasma with various platelet agonists such as epinephrine, ADP, collagen, arachidonic acid (AA), and ristocetin. According to the ISTH guideline, “extension panel” of platelet agonists can be used, such as thrombin receptor (PAR1) activating peptide (PAR1-AP), TRAP-6, and thromboxane A2 mimetic, U46619 [9, 20]. LTA measures changes in optical density or turbidity induced by agonists in PRP [21]. The parameters measured include the presence of shape change, length of the lag phase, rate of platelet aggregation, maximal percent of aggregation, percent aggregation at the end of the monitoring period, deaggregation, and the presence of a ‘secondary wave’ induced by epinephrine [20, 21]. Impaired platelet aggregation with more than one agonist suggests IPDs (Table 2, Fig. 2) [9]. The absence of aggregation of all agonists, except ristocetin, indicates Glanzmann thrombasthenia (GT) [4]. In contrast, the absence of a response to ristocetin suggests BSS [4]. A markedly decreased or absent aggregation of ADP suggests a P2Y12 receptor abnormality [4]. Decreased aggregation response to collagen and the absence of secondary waves to epinephrine and ADP point to platelet storage pool defects [4, 11]. Increased platelet aggregation in response to low concentrations of ristocetin is compatible with platelet-type vWD, which is due to a gain-of-function phenotype of platelet GPIbα and has an increased avidity for vWF, leading to the binding of the largest vWF multimers to resting platelets and their clearance from the circulation [4, 8, 11]. Scott syndrome should be considered in patients with mucocutaneous bleeding but normal LTA results [4], but LTA results may be normal or only slightly defective in some mild platelet function abnormalities, particularly dense granule deficiency. Therefore, normal LTA results alone do not entirely exclude IPDs [11, 22]. In addition, it should be noted that abnormal aggregation patterns to epinephrine are commonly observed even in non-IPD cases; therefore, further diagnostic processes should be considered if other abnormalities, strong clinical suspicion, or both are present [9]. The type of abnormal aggregation pattern in the LTA determines which workup will be required for the next step.
Although LTA is considered the gold standard for evaluating platelet function, it requires fresh blood and relatively large volumes of blood (20–50 mL) [23]. Therefore, it is challenging to implement in children with suspected IPDs. In addition, LTA is a time-consuming and technically complicated test because many pre-analytical and analytical variables can affect the results; therefore, it should be carefully controlled by expert laboratory personnel [20, 24]. Therefore, LTA cannot be routinely performed in many laboratories. In 2015, the ISTH published recommendations for performing LTA to solve standardization concerns [20]. Abnormal LTA results should be rechecked by performing the test on a second sample to exclude the effect of pre-analytical variables [11].
Whole-blood aggregometry (WBA) is an alternative method for measuring platelet aggregation. It measures the change in electrical resistance between two electrodes immersed in a whole blood sample, which results from the adhesion of platelets to the electrodes and subsequent platelet aggregation [25]. It overcomes the issue of sample volume; therefore, it can be used to evaluate IPDs in pediatric patients [11]. However, it cannot provide additional important information, such as changes in platelet shape, the presence of secondary waves, or deaggretation [20]. In addition, reproducibility, sensitivity, and specificity have not yet been established, the correlation with LTA is poor, and its ability to predict clinical outcomes is debatable [8, 11].
The use of the platelet function analyzer-100/200 for screening abnormal platelet function is not recommended due to the lack of reproducibility and low sensitivity and specificity for IPDs [9, 11]. Prolonged closure time (CT) cannot discriminate between vWD and other IPDs, and normal CT does not rule out mild platelet function disorders [11]. In addition, the results can be affected by low hematocrit and platelet counts; thus, they are not reliable in severe thrombocytopenic patients suspected with IPDs [11, 26].
Platelet secretion assessment
Impairment of granule release can be assessed by lumi-aggregometry, luminometry, high-performance liquid chromatography, ELISA, or flow cytometry [8, 9]. It should be possible to check for ADP/ATP release and at least a marker of α-granules [9]. If a secretion defect is identified, the next step is to measure the granule content [9].
Flow cytometry
Platelet glycoprotein (GP) expression can be assessed by flow cytometry using antibodies against GPIIb (CD41), GPIIIa (CD61), GPIb (CD42b), and GPIX (CD42a) [9, 11]. The GPIIb/IIIa activation epitope (PAC-1) can be used on activated platelets [9]. Defects in glycoprotein expression can indicate well-defined IPDs [9]. For example, reduced GPIIb/IIIa expression confirms the diagnosis of GT [8, 9, 11]. The defective expression of GPIb/IX is characteristic of BSS [27].
Other tests
Transmission electron microscopy (TEM) provides information on platelet structure related to platelet function, such as the number of α- and δ-granules, as well as identification of structural alterations that can be diagnostic for certain IPDs (Fig. 1C, D) [9, 11]. In addition, the clot retraction assay can assess the impairment of clot retraction, which can be found in GT or Stormorken syndrome [9].
Genetic analysis
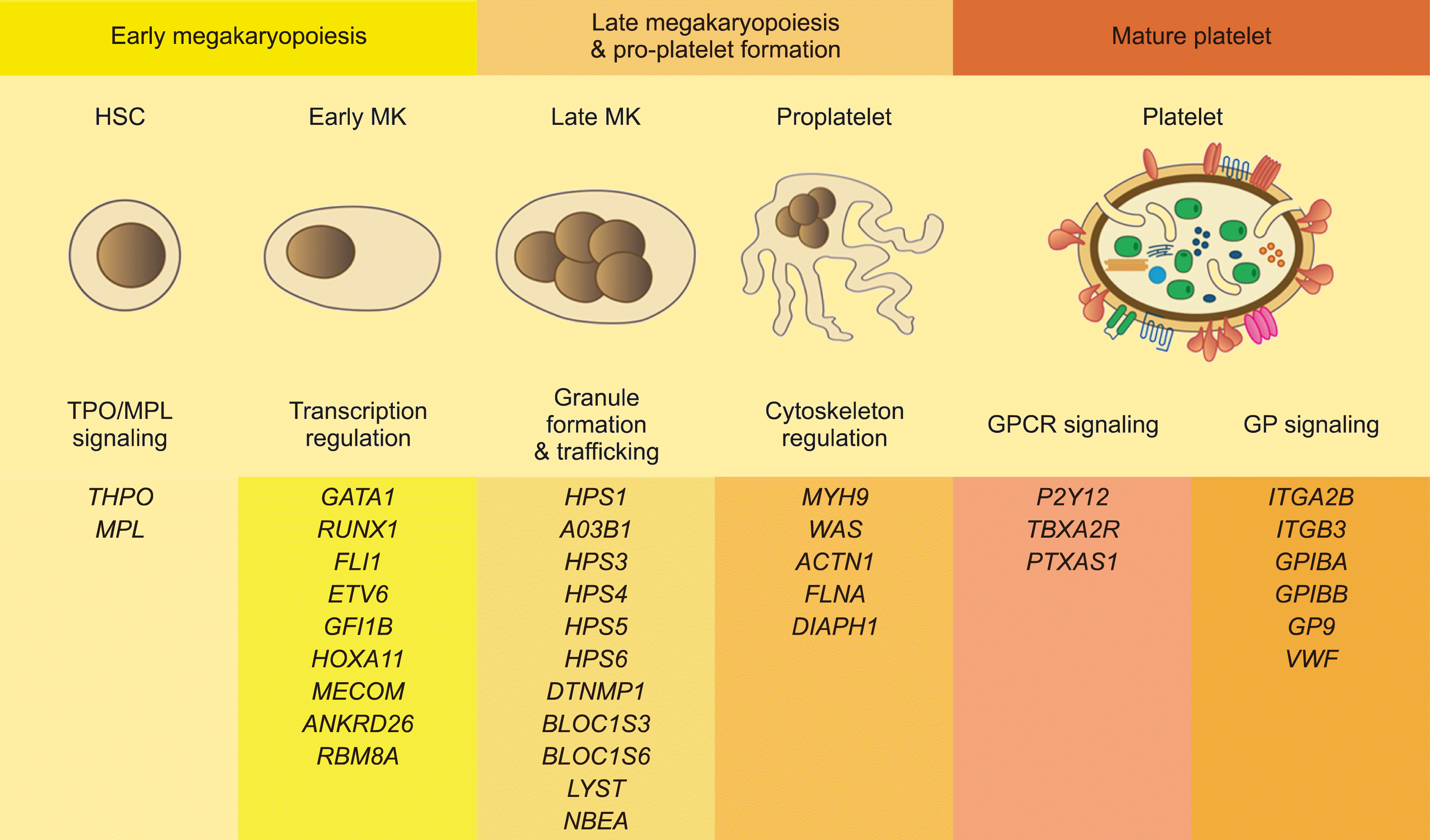
Defects in genes related to various steps during megakaryopoiesis often result in IPDs [1]. At least 51 genes associated with IPDs have been identified (Fig. 3) [1, 5, 7].
Genetic testing aims to determine the precise cause of IPDs, especially when other laboratory tests are not informative, confirm a diagnosis of IPDs suggested by the above-mentioned laboratory tests, and guide optimal treatment [2, 11]. In addition, molecular analysis can provide information on the disease course, such as IPDs associated with myelofibrosis, lung fibrosis, and malignancy [2].
Sanger sequencing of specifically targeted genes is the most widely used molecular analysis method. It is useful when the phenotype and clinical and laboratory findings are diagnostic for a monogenic disorder, for example, the analysis of MYH9 in a patient with macrothrombocytopenia and Döhle bodies (Fig. 1A) [11]. In addition, it can be effective if there is an affected family member with confirmed IPDs and the specific familial genetic variant is known [11]. The diagnosis of IPDs can be confirmed through targeted familial sequencing tests, and carrier status can be excluded [11].
Next-generation sequencing (NGS) can effectively detect sequence variants and small insertions or deletions effectively [11]. Gene panels for diagnosing IPDs have been reported, which include the most commonly affected genes [5, 10, 28, 29]. When a patient shows a non-specific phenotype but has been confirmed to have thrombocytopenia, platelet dysfunction, or both, disease-causing variants can be found through NGS using gene panels [11].
Array comparative genomic hybridization or multiplex ligation-dependent probe amplification can be performed to confirm copy number variants [11].
Genetic testing requires a relatively small amount of blood; therefore, diagnosis in children is possible [5]. Fresh platelets required in functional tests are unstable, but on the contrary, DNA used in genetic testing has the advantage of being stable [5].
The interpretation of genetic test results requires caution. It should be interpreted considering the clinical context and whether it is reported as a disease mutation in the database to avoid the risk of overinterpretation [5, 11]. The specificity of a clinical phenotype is the most critical information for the interpretation of genetic data and determining clinical significance [5, 11]. In addition, ethical issues, in particular, the possible identification of variants that lead to germline predisposition to malignancy (ANKRD26, ETV6, RUNX1) that require transparency, should be considered during genetic counseling because they are associated with a much higher psychological burden than expected in the context of evaluating mild to moderate IPDs [5, 11].
DIAGNOSTIC ALGORITHM FOR IPDS
Fig. 4 shows the diagnostic approach algorithm for IPDs. If a patient has bleeding manifestations typical of IPDs, specific features suggesting syndromic IPDs, or both, preliminary laboratory investigations, including CBC, PB smear, routine coagulation studies, and screening tests for vWD, should be started [4, 9]. The first step was to check for thrombocytopenia. When thrombocytopenia occurs, platelet size provides important information for differentiating some IPDs. If the patient shows macrothrombocytopenia, platelet-type vWD, BSS, MYH9-related disorders, and GPS should be considered after excluding ITP [4]. For BSS diagnosis, platelet surface GPIb/IX analysis using flow cytometry is required [4]. Genetic analysis of GPIBα, GPIBβ, and GPIX can also aid in the genetic counseling of family members [4, 27]. In addition to macrothrombocytopenia, personal andfamily history of hearing loss, renal dysfunction, cataracts, and Döhle-like bodies in neutrophils on the PB smear suggest MYH9-related disorders [8]. Genetic analysis of MYH9 confirmed the diagnosis. GPS typically shows large pale platelets on the PB smear, decreased platelet aggregation pattern with collagen, and absence of α-granules on TEM [4]. Platelet-type vWD results from the increased affinity of vWF for GPIbα owing to mutations in the Al domain of vWF [8]. Mildly decreased vWF with a disproportional decrease in high molecular weight multimers and enhanced platelet aggregation with ristocetin in LTA are characteristic findings [8].
When microthrombocytopenia occurs, WAS and XLT are possible diagnoses [4]. Further workup for diagnosing WAS includes bone marrow and immune function tests to identify immunodeficiencies [4]. IPDs that manifest thrombocytopenia with normal platelet size include congenital amegakaryocytic thrombocytopenia (CAMT), amegakaryocytic thrombocytopenia with radioulnar synostosis (ATRUS), familial platelet disorder with predisposition to acute myeloid leukemia (FPD/AML), thrombocytopenia with absent radii (TAR), and GATA-1 mutation of X-linked thrombocytopenia with thalassemia [4]. This finding may indicate abnormalities in megakaryopoiesis. Bone marrow studies and plasma thrombopoietin (TPO) level studies can help differentiate these disorders from acquired thrombocytopenia [4]. Some IPDs, such as ATRUS and TAR, show characteristic body abnormalities, and CAMT can be confirmed by TPO-receptor gene analysis [4].
If the patient showed a normal platelet count, the next step was performing LTA to confirm congenital platelet function disorders. Some IPDs present with characteristic abnormal patterns of LTA. The diagnostic hallmark of GT is the lack or severe defect in platelet aggregation induced by all agonists, except ristocetin. There was no impairment of ristocetin-induced platelet aggregation [8, 30]. Decreased platelet GPIIb/IIIa levels determined by flow cytometry can also lead to the diagnosis of GT [8]. In addition, mutations in ITGA2B and ITGB3 have been found in GT [30]. Decreased or absent aggregation of ADP suggests a P2Y12 receptor defect if the drug’s effect is excluded through careful medical history taking [4, 8]. Flow cytometric analysis of the surface P2Y12 receptor or molecular analysis of the P2Y12 gene can confirm the diagnosis [4]. α- or δ- granule deficiency (storage pool disease) is suspected when there is a defective secondary aggregation of ADP and epinephrine, and decreased aggregation of collagen [4, 8]. ATP or serotonin release assays can confirm the diagnosis [4, 8].
CONCLUSION
IPDs should be considered when a patent shows mucocutaneous bleeding in addition to ITP or acquired hemorrhagic disease. In patients with a family history suggestive of a hereditary feature or patients with significant bleeding histories, a comprehensive evaluation for the presence of IPDs should be started. Basic laboratory tests, including CBC and routine coagulation tests, are widely available, and careful interpretation of the results is important because it provides significant clues for determining the next testing step. A stepwise approach using platelet function tests and flow cytometric analysis is essential for diagnosing IPDs, although these are available in some specialized laboratories. Molecular analysis has become more popular nowadays and can help in accurate diagnosis and optimal treatment when appropriate genetic counseling is provided.

 XML Download
XML Download