

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The gastric mucosa is exposed to various intrinsic and extrinsic factors that can induce gastric mucosal damage. Gastric mucosal damage induced by a range of causes can lead to multiple pathologies, such as gastritis or gastric ulcers.1 Gastritis is one of the most common digestive tract diseases that can cause dyspepsia, abdominal pain, nausea, and vomiting.2
The known causes of gastric mucosal damage include gastric acid, pepsin, Helicobacter pylori, ethanol, and NSAIDs.1,3,4 These last two are common causes of gastritis. Ethanol damages the gastric mucosal barrier and induces the secretion of inflammatory cytokines, leading to the development of gastritis.5 NSAIDs, which inhibit mucosal prostaglandin production, can cause gastroduodenal mucosal damage.6
Tauroursodeoxycholic acid (TUDCA) is a conjugate of taurine and ursodeoxycholic acid (UDCA).7 TUDCA was reported to inhibit the apoptosis of hepatocytes.8 In an animal colitis model, TUDCA inhibited experimental colitis by preventing intestinal epithelial cell death.7 Recently, TUDCA attenuated colitis-associated tumorigenesis in a mouse model.9 In a study with cultured rabbit gastric cells, TUDCA had protective effects.10 Moreover, TUDCA had a gastric protective effect in an amphibian model.11 On the other hand, the pathophysiology and mechanism for protecting gastric epithelial cells by TUDCA are unclear. In this study, we aimed to elucidate that TUDCA has inhibitive effects on the nuclear factor kappa B (NF-κB) signaling in gastric epithelial cells. We also aimed to clarify the protective effects of TUDCA in a mouse gastritis model induced by ethanol and NSAIDs, and to compare the effects of TUDCA and UDCA.
Go to : 
SUBJECTS AND METHODS
1. Inhibitory effect of TUDCA on NF-κB signaling in gastric epithelial cells
1) Cell culture and TUDCA administration
The human gastric cell line MKN-45 was purchased from the Korean Cell Line Bank (Korean Cell Line Research Foundation, Cancer Research Institute, Seoul National University College of Medicine, Seoul, Korea). The MKN-45 cells were cultured in Roswell Park Memorial Institute medium 1640 (Welgene, Daegu, Korea) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin, as described elsewhere.12 The cultured MKN-45 cells were pretreated for 24 hours with various concentrations of TUDCA (Daewoong Pharmaceutical, Seoul, Korea) or with phosphate-buffered saline (PBS) and stimulated with tumor necrosis factor (TNF)-α (Sigma-Aldrich, St. Louis, MO, USA) to activate NF-κB signaling.
2) RNA extraction and real-time reverse transcription-PCR (RT-PCR)
The cellular RNA was extracted from MKN-45 cells using TRIzol reagent (GIBCO, Gaithersburg, MD, USA). Subsequently, 1 μg of the extracted RNA was reverse-transcribed and amplified using LightCycler 480 DNA SYBR Green I Master (Roche Applied Science, Penzberg, Germany) and LightCycler 480 II (Roche Diagnostics Ltd., Rotkreuz, Switzerland), as described in a previous study.13
3) Electrophoretic mobility shift assay (EMSA)
Pretreated MKN-45 cells were stimulated with TNF-α for 1 hour. The DNA binding activity of NF-κB was detected by EMSA analysis using a commercial kit according to the manufacturer’s instructions (Promega, Madison, WI, USA), as described elsewhere.13 A total of 5 μg of nuclear extract were incubated for 20 minutes at room temperature with a γ32P-labeled oligonucleotide probe, corresponding to the consensus NF-κB-binding site. The bound and free DNAs were separated on 5% non-denaturing polyacrylamide gels.
4) Immunoblot assay
MKN-45 cells pretreated with various TUDCA concentrations were stimulated with TNF-α for 1 hour. Immunoblot analysis for IκBα, phospho-IκBα, and β-actin was performed as described elsewhere.14 MKN-45 cells were washed with ice-cold PBS, and nuclear proteins were extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham, MA, USA). The protein concentrations in the lysates were determined using a Bradford assay. Fifty micrograms of cytoplasmic protein per lane were size-fractionated on a 12% polyacrylamide minigel and transferred to a nitrocellulose membrane (pore size, 0.45 μm). Specific proteins were detected using anti-IκBα, anti-phospho-IκBα, and anti-β-actin primary antibodies (Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Peroxidase-conjugated mouse IgG antibody was used as a secondary antibody. The target proteins were detected by a Luminescent Image Analyzer LAS 4000 (FujiFilm, Tokyo, Japan).
2. Ethanol-induced gastritis and the effect of pretreatment with TUDCA/UDCA
1) Mice
Procedures involving mice were reviewed and approved by the Institutional Animal Care and Use Committee of Seoul National University Boramae Medical Center (IACUC No. 2013-0002). The experimental procedures were performed according to the National Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Mice (C57BL/6, male, 6-7 weeks old) were purchased from Orient (Seongnam, Korea). The mice were given ad libitum access to water and standard rodent food until they reached the desired age (7-8 weeks) and body weight (18-20g). The mice were held under a 12 hours: 12 hours light/dark cycle and specific pathogen-free conditions. The sample size was calculated using ‘resource equation method’ described previously.15,16 The error degrees of freedom were calculated to be 20 with five mice per group, which means that the sample size of this study was appropriate according to the ‘resource equation method’.
2) Induction of ethanol-induced gastritis and pretreatment with TUDCA/UDCA
Twenty-five mice were divided into five groups using a block randomization method as follows (n=5 per group). The five groups are as follows: Group 1, negative control group; Group 2, ethanol (5 g/kg) group after pretreatment with PBS; Group 3, ethanol (5 g/kg) group after pretreatment with TUDCA (50 mg/kg); Group 4, ethanol (5 g/kg) group after pretreatment with TUDCA (250 mg/kg); Group 5, ethanol (5 g/kg) group after pretreatment with UDCA (50 mg/kg). The mice assigned to the negative control group received filtered water. TUDCA/UDCA was dissolved in PBS and administered 1 hour before ethanol administration via an oral gavage (Fig. 1A).
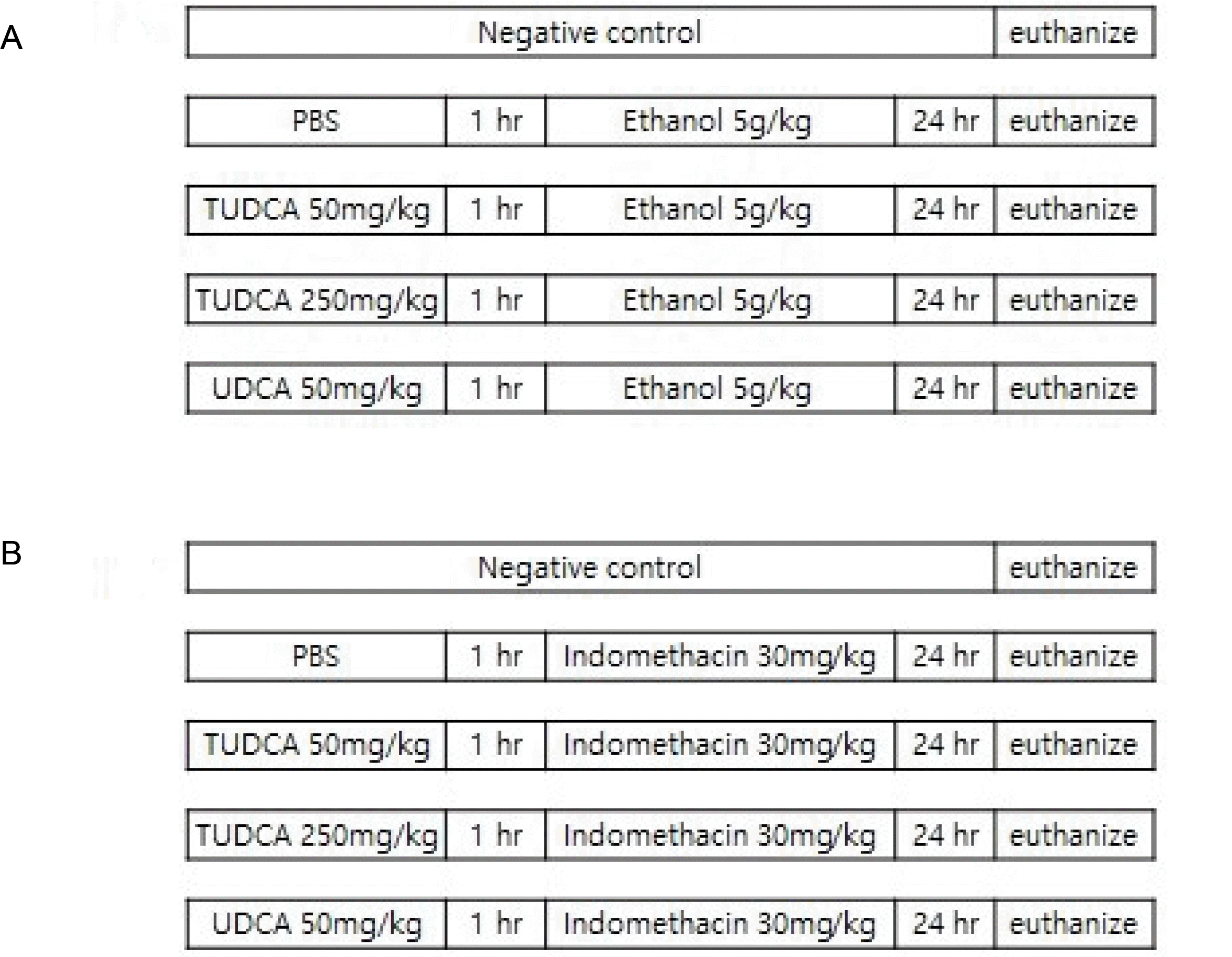 | Fig. 1Experimental protocol. (A) Ethanol-induced gastritis model (n=5 for each group). Group 1, negative control group, received filtered water; Group 2, ethanol (5 g/kg) after a pretreatment with PBS; Group 3, ethanol (5 g/kg) after a pretreatment with TUDCA (50 mg/kg); Group 4, ethanol (5 g/kg) after pretreatment with TUDCA (250 mg/kg); and Group 5, ethanol (5 g/kg) after pretreatment with UDCA (50 mg/kg). TUDCA or UDCA was dissolved in PBS and administered one hour before ethanol administration via oral gavage. (B) Indomethacin-induced gastritis model (n=5 for each group). Group 1, negative control group received filtered water; Group 2, indomethacin (30 mg/kg in 5% NaHCO3) after a pretreatment with PBS; Group 3, indomethacin (30 mg/kg in 5% NaHCO3) after a pretreatment with TUDCA (50 mg/kg); Group 4, indomethacin (30 mg/kg in 5% NaHCO3) after a pretreatment with TUDCA (250 mg/kg); and Group 5, indomethacin (30 mg/kg in 5% NaHCO3) after a pretreatment with UDCA (50 mg/kg). TUDCA or UDCA was dissolved in PBS and administered one hour before indomethacin administration via oral gavage. PBS, phosphate-buffered saline; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid.
|
3) Macroscopic scores
Twenty-four hours after ethanol administration, the mice were anesthetized deeply with isoflurane inhalation and euthanized for subsequent extraction of their stomachs.17 Possible confounders, such as the order of treatments and measurements, were not controlled in the current study. The stomach was cut longitudinally along the greater curvature, washed with PBS, and its gross appearance was evaluated.12 Five mice per group were included in the macroscopic and microscopic analyses without exclusion. A macroscopic evaluation was performed quantitatively by one researcher blinded to the group assignment and the conduct of the experiment. The attributed scores were as follows: 0, normal; 0.5, light local reddening; 1, general redness or small hemorrhage (<1 mm); 2, large hemorrhage (≥1 mm); 3, small ulcer (<2 mm); 4: large ulcer (≥2 mm); and 5, perforated ulcer.
4) Microscopic scores
The removed tissues were fixed in 10% buffered formalin and embedded in paraffin. H&E was performed, and histological quantification was conducted using a scoring system. Two pathologists blinded to the group assignment performed the histologic evaluation quantitatively. Microscopic analyses were conducted, and the scores in each group were compared.12 The microscopic mucosal damage scores were as follows: 0, normal; 1, slight damage of surface gastric mucosa or damage of just two or three glandular cells in the upper mucosal layer; 2, damage greater than that of score 1 and involving <50% of the thickness of the gastric mucosa; 3, damage involving >50% of the thickness of the gastric mucosa. The attributed mucosal damage area scores were: 1, <10% of the gastric mucosa area; 2, 10-25% of the gastric mucosa area; 3, 25-50% of the gastric mucosa area; and 4, >50% of the gastric mucosa area.
The mucosal damage index was calculated as follows:
Mucosal damage index=(microscopic mucosal damage score)×(mucosal damage area score)
3. NSAID-induced gastritis and effect of pretreatment with TUDCA/UDCA
1) Mice
The mice (C57BL/6, male, 6-7 weeks old) were purchased from Orient (Seongnam, Korea). The mice were given access to water and standard rodent food ad libitum until they reached the desired age (7-8 weeks) and body weight (18-20 g). The mice were held under a 12 hours: 12 hours light/dark cycle and specific pathogen-free conditions.
2) Induction of indomethacin-induced gastritis and pretreatment with TUDCA/UDCA
Twenty-five mice were divided into five groups using a block randomization method as follows (n=5 per group): Group 1, negative control group; Group 2, indomethacin (30 mg/kg in 5% NaHCO3) group after pretreatment with PBS; Group 3, indomethacin (30 mg/kg in 5% NaHCO3) group after pretreatment with TUDCA (50 mg/kg); Group 4, indomethacin (30 mg/kg in 5% NaHCO3) group after pretreatment with TUDCA (250 mg/kg); Group 5, indomethacin (30 mg/kg in 5% NaHCO3) group after pretreatment with UDCA (50 mg/kg). The mice were assigned to the negative control group that had received filtered water. TUDCA/UDCA was dissolved in PBS and administered 1 hour before indomethacin administration via an oral gavage (Fig. 1B).
3) Macroscopic scores
Twenty-four hours after indomethacin administration, the mice were deeply anesthetized with isoflurane inhalation and euthanized for subsequent extraction of their stomachs.17 The stomach was cut longitudinally along the greater curvature, washed with PBS, and the gross appearance was evaluated.12 Five mice per group were included in the macroscopic and microscopic analyses without exclusion. A macroscopic evaluation was performed quantitatively by one researcher blinded to the group assignment and the conduct of the experiment. The attributed scores were similar to the ethanol-induced gastritis model.
4) Microscopic scores
The removed tissues were fixed in 10% buffered formalin and embedded in paraffin. H&E was performed, and the histological quantification was performed using a scoring system. Two pathologists blinded to the group assignment performed the histologic evaluation quantitatively. Microscopic analyses were performed, and the scores were compared.12 The attributed microscopic mucosal damage scores were similar to the ethanol-induced gastritis model.
4. Statistical analysis
Statistical analysis was performed using SPSS Statistics v ersion 26.0 (IBM, Armonk, NY, USA). The differences between the groups were estimated using the Kruskal-Wallis test and a Mann-Whitney U test. p-values <0.05 were considered significant.
Go to :
RESULTS
1. Inhibitory effect of TUDCA on NF-κB signaling in gastric epithelial cells
1) RT-PCR
Human gastric MKN-45 cells were cultured and pretreated with TUDCA (50 and 250 mg/kg) for 24 hours and stimulated with TNF-α for 1 hour. The RNA was extracted from the gastric cell lines, reverse-transcribed, and amplified using the specific primers for human interleukin (IL)-1α and IL-1β. The RT-PCR results showed that TNF-α-induced upregulation of IL-1α mRNA expression was reduced significantly by the TUDCA pretreatment (Fig. 2).
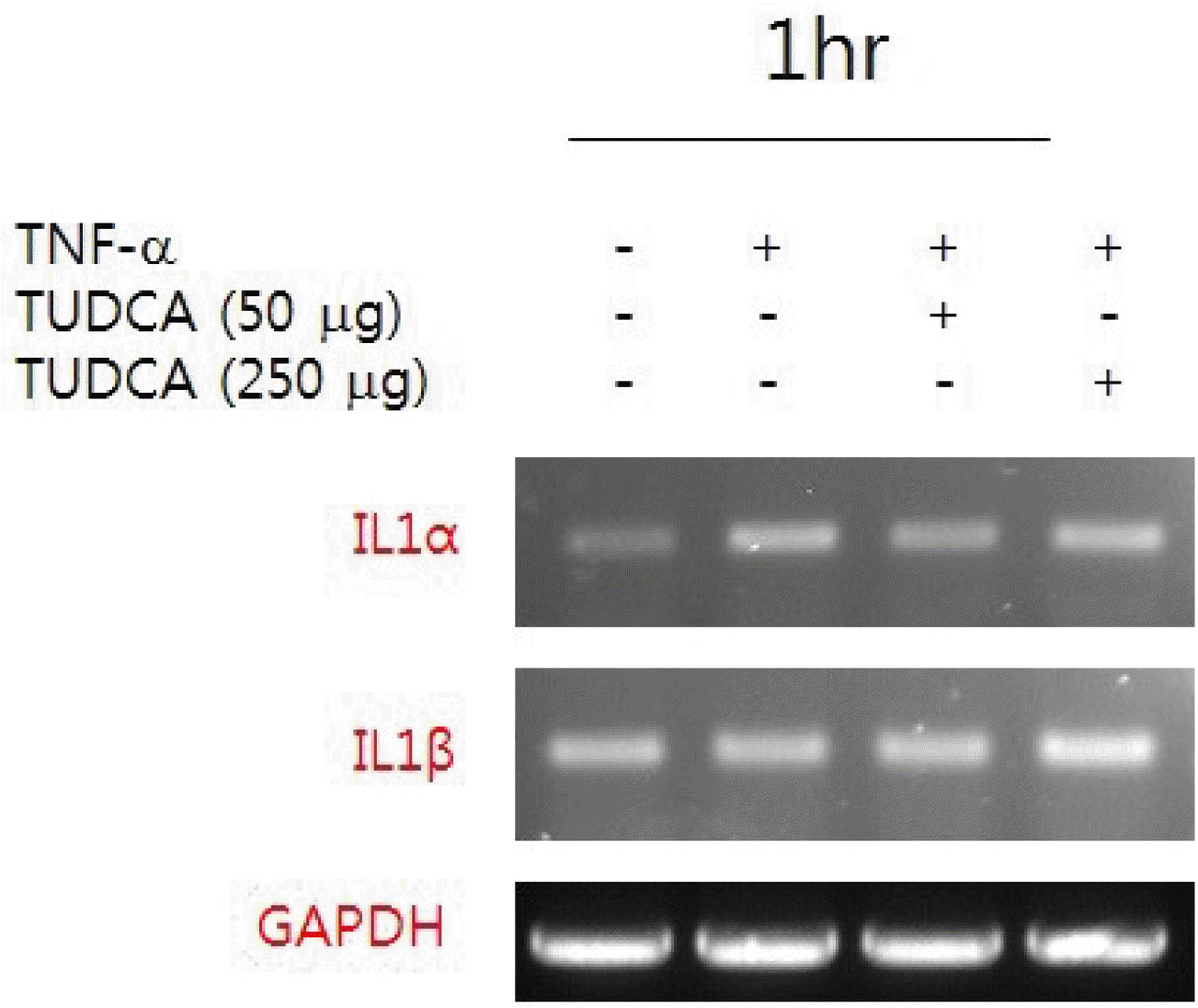
2) EMSA
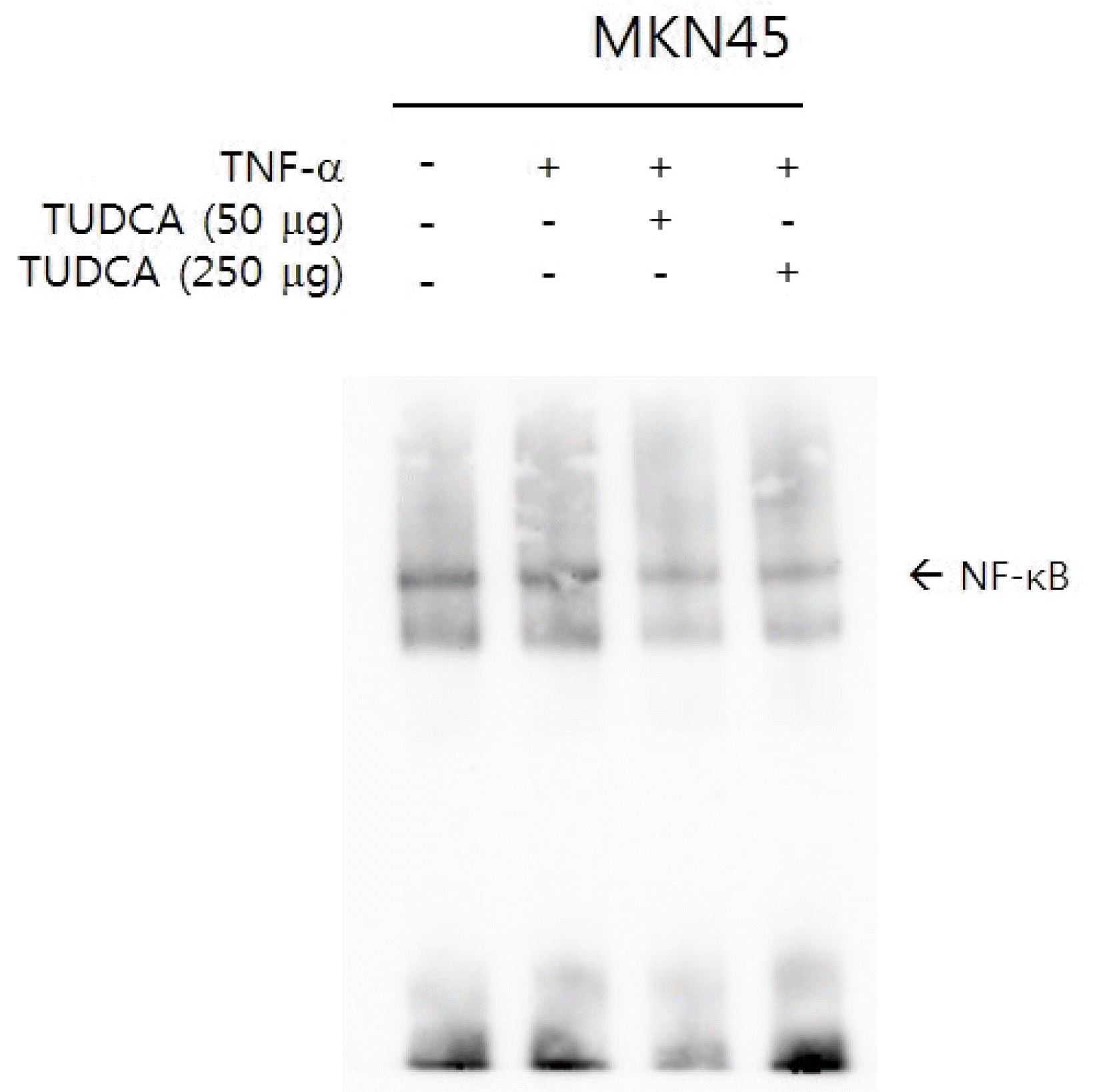
3) Immunoblot assay
The pretreatment of MKN-45 cells with various concentrations of TUDCA suppressed TNF-α-induced IκBα phosphorylation (Fig. 4A). The results of densitometry analyses show the quantification data in graphs (Fig. 4B, C).
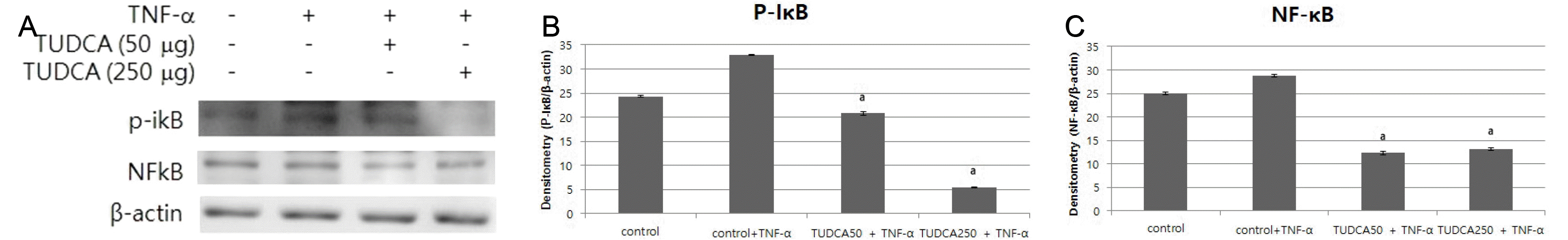 | Fig. 4(A) Pretreatment of MKN-45 cells with various concentrations of TUDCA suppressed TNF-α-induced IκBα phosphorylation. (B) Pretreatment of MKN-45 cells with TUDCA (50 and 250 μg/mL) suppressed IκBα phosphorylation. (C) Pretreatment of MKN-45 cells with TUDCA (50 and 250 μg/mL) suppressed NF-κB signaling. TUDCA, tauroursodeoxycholic acid; TNF, tumor necrosis factor. ap<0.05 compared with TNF-α alone.
|
2. Ethanol-induced gastritis and the effect of pretreatment with TUDCA/UDCA
1) Macroscopic scores
Gastritis was induced by the administration of ethanol. The pretreatment with 50 mg/kg and 250 mg/kg TUDCA attenuated the severity of gastritis (p=0.049 and p=0.005, respectively). On the other hand, pretreatment with 50 mg/kg UDCA did not (Fig. 5).
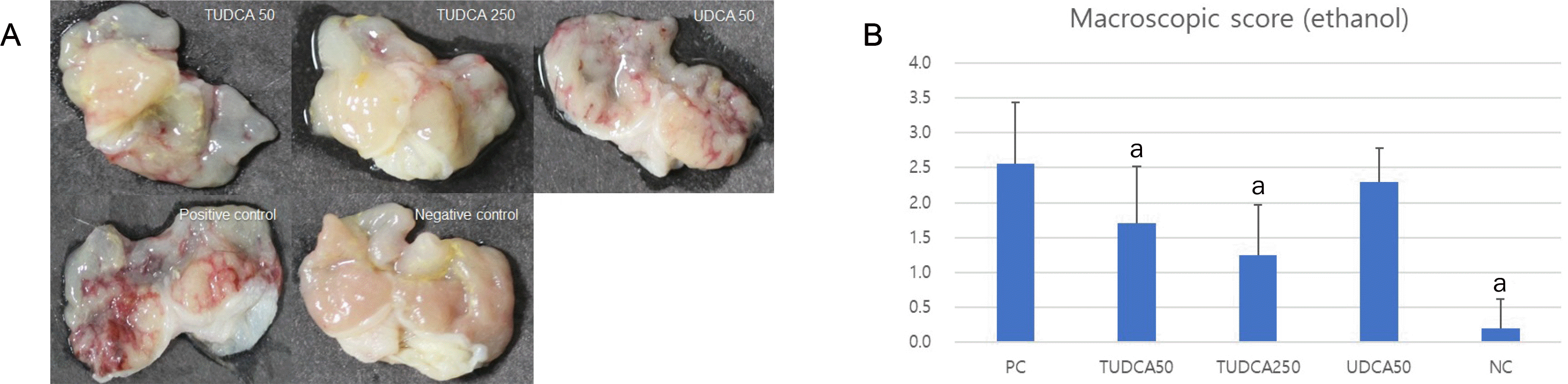 | Fig. 5Pretreatment with 50 and 250 mg/kg TUDCA significantly attenuated the severity of ethanol-induced gastritis. (A) Macroscopic findings of ethanol-induced gastritis. (B) Macroscopic scores of ethanol-induced gastritis. PC, positive control; NC, negative control; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid; TNF, tumor necrosis factor. ap<0.05 compared with TNF-α alone.
|
2) Microscopic scores
Gastritis was induced by the administration of ethanol. Pretreatment with TUDCA 250 mg/kg attenuated the severity of gastritis (p=0.009), but pretreatment with TUDCA 50 mg/kg and UDCA 50 mg/kg did not (Fig. 6).
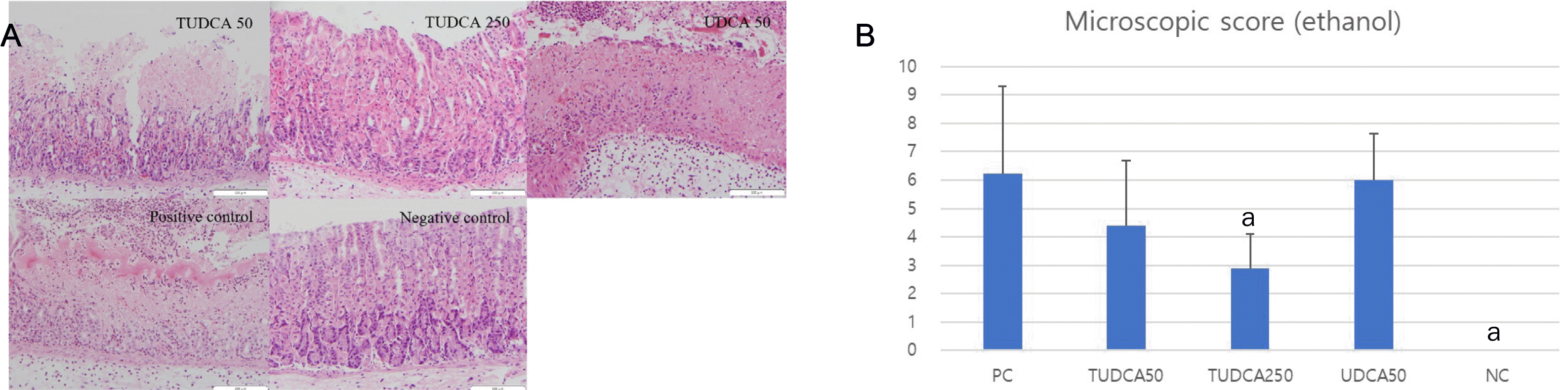 | Fig. 6Pretreatment with 250 mg/kg TUDCA significantly attenuated the severity of ethanol-induced gastritis. (A) Microscopic findings of ethanol-induced gastritis (hematoxylin and eosin, ×200). (B) Microscopic scores of ethanol-induced gastritis. PC, positive control; NC, negative control; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid; TNF, tumor necrosis factor. ap<0.05 compared with TNF-α alone.
|
3. NSAID-induced gastritis and effect of pretreatment with TUDCA/UDCA
1) Macroscopic scores
Gastritis was induced by the administration of indomethacin. The pretreatment with TUDCA 250 mg/kg attenuated the severity of gastritis (p=0.012), but the pretreatment with TUDCA 50 mg/kg and UDCA 50 mg/kg did not (Fig. 7).
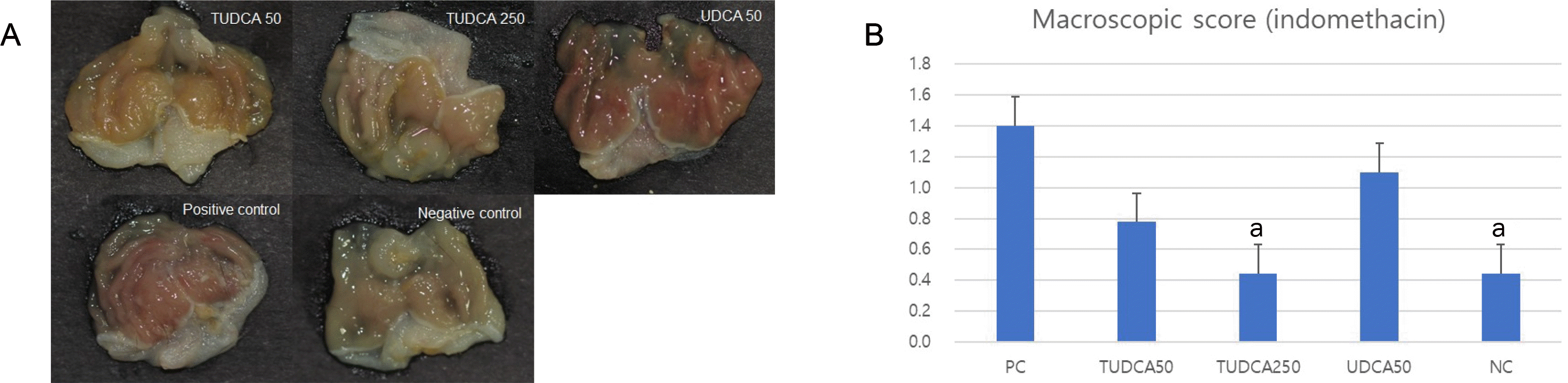 | Fig. 7Pretreatment with 250 mg/kg TUDCA significantly attenuated the severity of indomethacin-induced gastritis. (A) Macroscopic findings of indomethacin-induced gastritis. (B) Macroscopic scores of indomethacin-induced gastritis. PC, positive control; NC, negative control; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid; TNF, tumor necrosis factor. ap<0.05 compared with TNF-α alone.
|
2) Microscopic scores
Gastritis was induced by the administration of indomethacin. The pretreatment with TUDCA 250 mg/kg attenuated the severity of gastritis (p=0.048). On the other hand, the pretreatment with TUDCA 50 mg/kg and UDCA 50 mg/kg did not (Fig. 8).
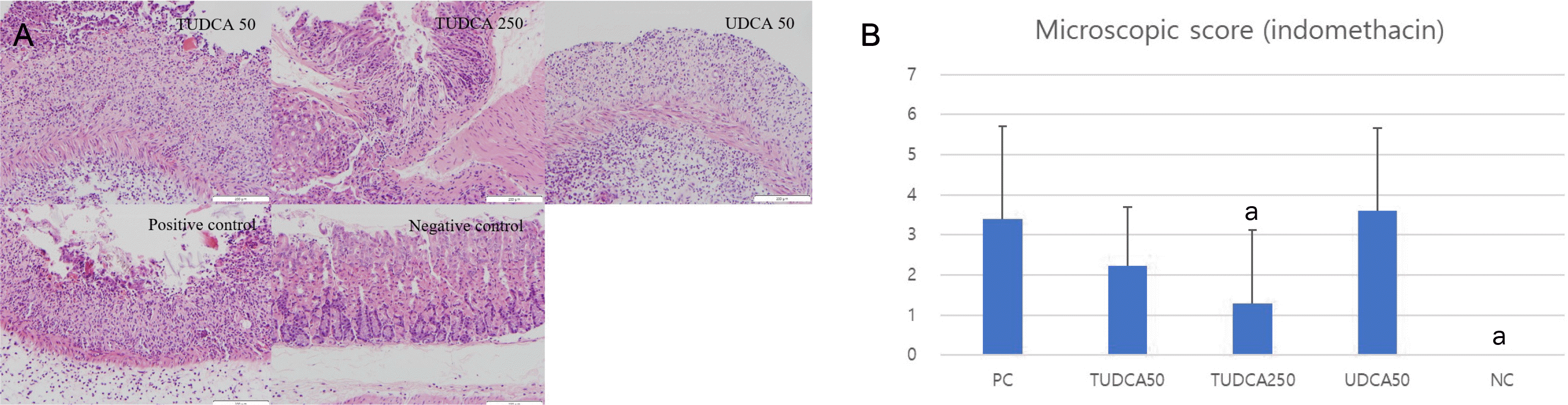 | Fig. 8Pretreatment with 250 mg/kg TUDCA significantly attenuated the severity of indomethacin-induced gastritis. (A) Microscopic findings of indomethacin-induced gastritis (hematoxylin and eosin, ×200). (B) Microscopic scores of indomethacin-induced gastritis. PC, positive control; NC, negative control; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid; TNF, tumor necrosis factor. ap<0.05 compared with TNF-α alone.
|
Go to :
DISCUSSION
In a series of studies, the persistent activation of NF-kB is a key factor in the development of chronic mucosal inflammation.18 In addition, endoplasmic reticulum stress is involved in the pathophysiology of inflammatory bowel diseases and is associated with NF-kB signaling.19 Recent colitis experimental studies showed that TUDCA inhibited colitis by preventing early intestinal epithelial cell death.7 In a study with cultured rabbit gastric cells, TUDCA exhibited protective effects against gastric mucosal damage.10 On the other hand, the pathophysiology and mechanism of protecting gastric epithelial cells by administrating TUDCA have not been elucidated. This study showed that TUDCA inhibited IL-1α signaling in gastric epithelial cells. The pretreatment with TUDCA inhibited the TNF-α-induced NF-κB DNA binding activity, and suppressed TNF-α-induced IκBα phosphorylation. To clarify the effects identified in the in vitro experiments, we performed our study in ethanol-and NSAID-induced gastritis animal models. The results showed that TUDCA attenuates ethanol-induced and NSAID-induced gastritis.
NF-κB activation plays a leading role in intestinal permeability20 and NF-κB signaling also plays a role in regulating inflammation, apoptosis, and tumorigenesis.21,22 Most NF-κBs exist as homodimers or heterodimers. When there is no stimulation, NF-κB is located in the cytoplasm, conjugated with IκBα, which prevents NFκB from shifting into the nucleus. On the other hand, with stimulation, such as viruses or TNF-α, IκBα is phosphorylated by IκB kinase and degraded, and NF-κ B migrates into the nucleus and enhances the expression of many pro-inflammatory genes.18 The present results showed that TUDCA suppressed TNF-α-induced IκBα phosphorylation and IL-1α signaling in gastric epithelial cells. This study is the first to show the protective effects of TUDCA in gastric epithelial cells and an animal model. The gastro-protective pathophysiology and mechanism of TUDCA were also studied.
Two gastritis mouse models with ethanol and NSAIDs were used to determine the gastro-protective effect. In the macroscopic assessment of the ethanol-induced gastritis model, the pretreatment with 50 and 250 mg/kg TUDCA showed a protective effect against ethanol-induced gastritis, but 50 mg/kg UDCA did not. These results are consistent with those of a previous study in which TUDCA rather than UDCA had protective effects against gastric cell damage.10 Ota et al.10 reported a TUDCA-induced increase in prostaglandin E2 production that facilitated its gastroprotective effects. In the microscopic assessment of ethanol-induced gastritis, the pretreatment with TUDCA 250 mg/kg had a protective effect against ethanol-induced gastritis, but 50 mg/kg TUDCA and 50 mg/kg UDCA did not. In the microscopic and macroscopic assessment of the NSAID-induced gastritis model, the pretreatment with 250 mg/kg TUDCA had a protective effect against NSAID-induced gastritis, but 50 mg/kg TUDCA and 50 mg/kg UDCA did not. These results are consistent with the previously reported dose-dependent gastroprotective effects of TUDCA.10
The main therapeutic principle of gastric mucosal inflammation is suppressing gastric acid secretion.23 Typical medications that inhibit gastric acid secretion are H2-receptor antagonists and proton pump inhibitors (PPI). On the other hand, H2-receptor antagonists develop rapid tolerance during therapy, and the withdrawal of H2-receptor antagonists can induce acid rebound.24,25 The long-term use of PPI is associated with various complications, such as pneumonia, Clostridium difficile infections, and gastric polyps.26-29 In patients with liver cirrhosis, the risk of spontaneous bacterial peritonitis and mortality increased with PPI use.30,31 In addition, the role of gastric acid is small in the gastric mucosal damage induced by ethanol because ethanol does not increase the gastrin level in the blood.32 Thus, PPIs are unlikely to be useful in treating gastric mucosal damage induced by ethanol. Therefore, in the present study, the aim was to develop a new drug to protect the gastric mucosa from ethanol-induced gastric damage. TUDCA has a protective effect against gastritis in ethanol-induced gastritis models, along with its mechanism that TUDCA inhibits NF-κB signaling in gastric epithelial cells. The results showed that TUDCA protects against gastritis in NSAID-induced gastritis models. Further studies will be needed to determine if TUDCA can be applied to gastritis models other than ethanol- or NSAID-induced models.
This study had some limitations. First, only one cell line was used for the cell study. Second, although the macroscopic and microscopic scores of the gastric mucosal damage were used to evaluate the protective effects of TUDCA in gastritis mouse models, the anti-inflammatory effects of TUDCA through NF-κB pathways or inflammatory cytokine expression were not evaluated in gastritis mouse models in the current study. On the other hand, this study clearly showed that TUDCA ameliorated gastric mucosal damage in the ethanol-and NSAID-induced gastritis mouse models.
In conclusion, TUDCA inhibited NF-κB signaling in gastric epithelial cells and ameliorated ethanol- and NSAID-induced gastritis in murine models. The study results suggest the possibility that TUDCA may be used to prevent gastritis induced by ethanol or NSAIDs, but more research is needed.
Go to :
 XML Download
XML Download