

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Carbon monoxide (CO), an important gasotransmitter endogenously produced via the degradation of heme by heme-oxygenases (HOs) [1], possesses similar biological and pharmacological properties to nitric oxide (NO) and hydrogen sulfide [2-4]. As active signaling molecules involved in a variety of physiological and pathological processes, these gasotransmitters have been the focus of increasing attention over the past decade [2-6]. CO has been previously regarded only as a highly toxic gas for its role in preventing O2 transport and delivery [7], and involvement in the inducement of cardiac dysfunction [8-10]. However, CO is now known to be a neurotransmitter, as well as a cardiovascular and immune regulator [2,5]. Numerous studies have shown that low concentrations of CO exhibit pleiotropic biological functions, including anti-apoptotic, cytoprotective, and anti-inflammatory effects [11-13]. Cardiovascular tissues in normal conditions in particular express very low levels of HO-1 (an inducible form of HO), which is upregulated following stress symptoms such as high temperature and ischemia/reperfusion (I/R) or in certain pathological conditions [4,14-16]. The HO/CO system has been reported as being responsible for the numerous positive effects in studies of cardiovascular [13] and lung disorders [6,17].
The cardiac hormone atrial natriuretic peptide (ANP) is released primarily from the cardiac atria and plays an important role in various physiological and pathological processes [18,19]. ANP decreases blood pressure by regulating extracellular fluid (ECF) volume and vasodilation, while exerting numerous beneficial effects on cardiovascular disorders and metabolic syndromes [20]. In addition, ANP levels serve as a potential biomarker for the diagnosis of the acute stages of heart failure. The factors that determine the amount of circulating ANP are intravascular volume change [21,22], solute concentration, and presence of several hormones [23,24]. Therefore, increased plasma ANP levels are an important trigger for the compensatory mechanisms of the cardiovascular system that respond to physiological and pathological conditions. The effects of CO vary across tissues, but involve the modulation of multiple pathways that contain NO, reactive oxygen species, and P38 MAP kinases [4]. What remains unknown are the details of the relationship between CO’s beneficial effects and ANP.
Several boron- or ruthenium-based compounds (carbon monoxide releasing molecules, CORMs) have been synthesized in the last decade. These exhibit pharmaceutical traits superior to those of gaseous CO by virtue of their chemistry and structure [25,26]. We performed this study using CORM and isolated perfused beating rat atria to determine the direct effect of CO on ANP secretion and the underlying signaling mechanisms involved.
METHODS
Animals
Male Sprague–Dawley rats (Dae Han Bio Link, Eumsung, Korea), weighing 200–250 g each, were housed in temperature-controlled rooms with a 12 h light–dark cycle. Animals were given free access to standard laboratory chow (5L79 Purina rat and mouse 18% chow; Charles River Laboratories Inc., Wilmington, MA, USA) and water. All experimental protocols conformed to the guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication number 85-23, revised 1996) and were approved by our institution.
Chemicals
ANP was purchased from Bachem Holding AG (Bubendorf, Switzerland). CORM-2, CORM-3, Hemin, zinc porphyrin (ZnPP), pinacidil, 5-hydroxydecanoate (5-HD), diltiazem, NG-nitro-L-arginine methyl ester (L-NAME), paxilline, wortamannin, LY-294002 (LY), and 1H-[1,2,4] oxadiazolo [4,3-a] quinoxalin-1-one (ODQ) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Preparation of perfused beating atria
Isolated perfused beating atria were prepared as previously described elsewhere [27,28]. In brief, hearts were rapidly excised from rats and a cannula was fixed into the left atria. The cannulated atria were fixed in an organ chamber, perfused with oxygenated 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffered saline (10 mM HEPES, 118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 25 mM NaHCO3, 10 mM glucose, and 0.1% bovine serum albumin) at 37°C, and then paced at 1.2 Hz (30 V, 0.4 ms). The intra-atrial pressure was recorded using a Power Lab (ML-820, ADInstruments Pvt. Ltd., Sydney, Australia); atrial pulse pressure (APP) is defined as the difference between the systolic (SAP) and diastolic atrial pressure (DAP). The pericardial buffer solution that contained [3H]-inulin (Amersham Biosciences, Uppsala, Sweden) and that was used for measuring the translocation of ECF was also oxygenated through silicone tube coils inside the organ chamber. After ANP secretion stabilization and ECF translocation for 80 min, atrial perfusate was collected at 2 min intervals over 50 min at 4°C.
The groups were divided as follows. Group 1 was comprised of the control atria (n = 10). After a 10 min control period, atria were high-stretched by increasing the height of the outflow catheter from 5 cmH2O to 7.5 cmH2O by connecting a 2.5 cm long catheter, and atrial perfusate was continuously collected. The loaded volume to atria during diastole was 736 µl. Group 2 was comprised of atria perfused with CORM-2. After a 10 min control period, atria were perfused with various doses of CORM-2 (10, 50, 100 µM; n = 10 for each group) with high stretch. Group 3 was comprised of atria perfused with CORM-3, HO activator (hemin), and HO inhibitor (ZnPP). After a 10 min control period, atria were perfused with CORM-3 (10, 50, 100 µM; n = 10 for each group), hemin (0.1, 1, 50 µM; n = 10 for each group), ZnPP (50 µM; n = 10 for each group) with high stretch. Group 4 was comprised of atria perfused with CORM-2 (50 µM) in the presence of regulators, including KATP channel activator (pinacidil, 50 µM), mitochondrial KATP channel blocker (5-hydroxydecanoic acid, 5-HD, 100 µM), L-type Ca2+ channel blocker (diltiazem, 50 µM), high-conductance Ca2+-activated K+ (BKCa) channel blocker (paxilline, 25 µM), nitric oxide synthase (NOS) inhibitor (L-NAME, 100 µM), PI3K inhibitor (wortamannin, 10 µM; LY-294002, 10 µM), and soluble guanylate cyclase (sGC) inhibitor (ODQ, 100 µM). Atria were perfused with a regulator 20 min before sample collection, and CORM-2 (50 µM, n = 10 for each group) was simultaneously perfused into the high-stretched atria after a 10 min control period. Group 5 was comprised of atria perfused with CORM-2 (10 nM, 50 µM) in a hypoxic condition, as opposed to a normoxic condition. Atria were perfused with oxygen-free HEPES buffered saline, and CORM-2 (10 nM or 50 µM, n = 7 for each group) was simultaneously perfused into the high-stretched atria after a 10 min control period.
Measurement of ANP concentration and ECF translocation
The ANP concentration in the perfusate was measured using a specific radioimmunoassay (RIA) method previously described elsewhere [21]. The ANP in the plasma and coronary effluent was extracted using Sep-Pak C18 cartridges, dried, and measured by RIA [29]. The intra- and inter-assay coefficients of variation were 6.3% (n = 9) and 7.8% (n = 11), respectively. The amount of secreted ANP was presented as ng of ANP/g atrial wet weight/min. The molar concentration of ANP released is determined by:
The radioactivity of [3H]-inulin in the perfusates and pericardial buffer solution was measured with a 2 ml Ultima Gold cocktail solution (PerkinElmer, Waltham, MA, USA) using a liquid scintillation system (Tri-Card 300C; Packard, Downers Grove, IL, USA). ECF translocation was calculated as follows [30,31]:
Statistical analysis
All values are expressed as means ± SEM. Differences in hemodynamic variables over time between the control and the treatment groups were assessed using ANOVA followed by Bonferroni’s multiple comparison test (Sigmaplot; Systat Sofeware, Inc., Erkrath, Germany). The significance level was set at p < 0.05.
RESULTS
Effects of CORM-2 on ANP secretion and atrial contractility
After a 10-min collection period, each dose of CORM-2 (10, 50, or 100 µΜ) or vehicle was perfused into high-stretched atria, and atrial pressure and ANP secretion were measured. Fig. 1A shows ANP secretion, SAP, DAP, and APP over time. There was no significant difference in basal values of ANP secretion and atrial pressure among groups in this study. When the atria were high-stretched by increasing the height of outflow catheter from 5 cmH2O to 7.5 cmH2O, ANP secretion was gradually increased from 4.52 ± 0.73 ng/min/g of atrial tissue (at 10 min) to 11.54 ± 2.42 ng/min/g of atrial tissue (at 50 min) (Fig. 1Aa). SAP, DAP, and APP were increased suddenly and the higher level was maintained throughout the experiment (Fig. 1Ab–d). When 10 µΜ of CORM-2 was perfused into the high-stretched atria, ANP secretion was increased from 6.81 ± 0.91 ng/min/g of atrial tissue (at 10 min) to 16.88 ± 2.41 ng/min/g of atrial tissue (at 50 min) (Fig. 1Aa). When the dose of CORM-2 was increased to 50 µΜ, ANP secretion was rose slightly from 5.20 ± 0.62 ng/min/g of atrial tissue (at 10 min) to 8.67 ± 0.57 ng/min/g of atrial tissue (at 50 min). However, 100 µΜ of CORM-2 decreased ANP secretion from 8.08 ± 0.49 ng/min/g of atrial tissue to 6.09 ± 1.40 ng/min/g of atrial tissue.
To quantitatively compare the effects of CORM-2 on ANP secretion and atrial pressure, the relative percent change between the mean of the first five control values (fraction no. 1 to 5) and the mean of the last five peak values (fraction no. 21 to 25) was calculated (Fig. 1A). The ANP secretion in atria perfused with 10 µΜ CORM-2 changed by 121.7 ± 14.6%, which was similar to that observed in the control atria (124.9 ± 17.6%) (Fig. 1Ba). However, changes in ANP secretion in atria perfused with 50 µΜ or 100 µΜ CORM-2 were 63.9 ± 12.1% and –29.1 ± 14.3%, respectively, which were significantly lower than the amount of secretion observed in control atria (124.9 ± 17.6%) and in the 10 µM CORM-2. There was a significant difference in ANP secretion induced by 10 µM CORM-2 and 100 µM CORM-2 (Fig. 1Ba). Changes in SAP, DAP, and APP after three doses of CORM-2 were no different from those observed in the control group (Fig. 1Bb–d).
Effects of CORM-3, hemin, and zinc protoporphyrin on ANP secretion and atrial contractility
The effects on ANP secretion of other CO-related chemicals—CORM-3, hemin (a HO activator), and ZnPP (a HO inhibitor)—were compared with CORM-2. The same doses of CORM-3 (10, 50, and 100 µM) as CORM-2 were used but changes in ANP secretion were not significantly different from those observed in the control atria (121.7 ± 14.5%, 120.7 ± 7.7%, 103.5 ± 14.5% vs. 124.8 ± 17.6%) (Fig. 2). Hemin 0.1 µM did not suppress ANP secretion, but higher doses of hemin (1 and 50 µM) markedly reduced ANP secretion (45.3 ± 1.6%, –2.6 ± 0.2% vs. 124.8 ± 17.6%) (Fig. 2A). No significant differences in APP were found among samples administered CORM-2, CORM-3, and hemin (Fig. 2B). The order of potency in suppressing ANP secretion was hemin > CORM-2 >> CORM-3. Interestingly, ZnPP (50 µM) did not cause any significant change in ANP secretion (Fig. 2Ad) or APP (Fig. 2Bd).
To compare the effect of ANP secretion, CORM-2 or hemin (0.1 nM to 100 µM) was perfused into isolated rat atria. As shown in Fig. 3, between 0.1 nM and 10 µM CORM-2 did not result in any significant effects on ANP secretion. Nor did 0.1 nM to 100 nM hemin. CORM-2 and hemin began to suppress ANP secretion once administered at levels of 50 µM and 1 µM, respectively. Hemin appears to be stronger to suppress ANP secretion compare to CORM-2.
Modulation of the suppressive effect of CORM-2 on high stretch-induced ANP secretion
Previous reports have indicated that the ability of CO to relax blood vessels occurs through cGMP-dependent and independent pathways [4]. To determine the mechanisms by which the suppressive effect of CORM-2 (50 µM) on high stretch-induced ANP secretion operate, atria were pretreated with either mitochondrial KATP channel blocker (5-HD), KATP channel opener (pinacidil), L-type Ca2+ channel blocker (diltiazem), BKCa channel blocker (paxilline), NOS inhibitor (L-NAME), sGC inhibitor (ODQ), or PI3K inhibitor (wortmannin, LY). 5-HD (500 µM) itself decreased ANP secretion (56.1 ± 8.9%, Fig. 4A) relative to the control group (124.9 ± 17.6%). Pretreatment with 5-HD blocked CORM-2-induced suppression of ANP secretion and concentration (Fig. 4). Pretreatment with pinacidil did not block CORM-2-induced suppression of ANP secretion (Fig. 4A), while pretreatment with paxilline was effective at blocking CORM-2-induced suppression of ANP secretion and concentration. ODQ blocked CORM-2-induced suppression of ANP concentration but not ANP secretion (Fig. 4D). Diltiazem, L-NAME, and PI3K inhibitors (LY, wortmannin) had no significant effect on CORM-2-induced suppression of ANP secretion (Fig. 4A).
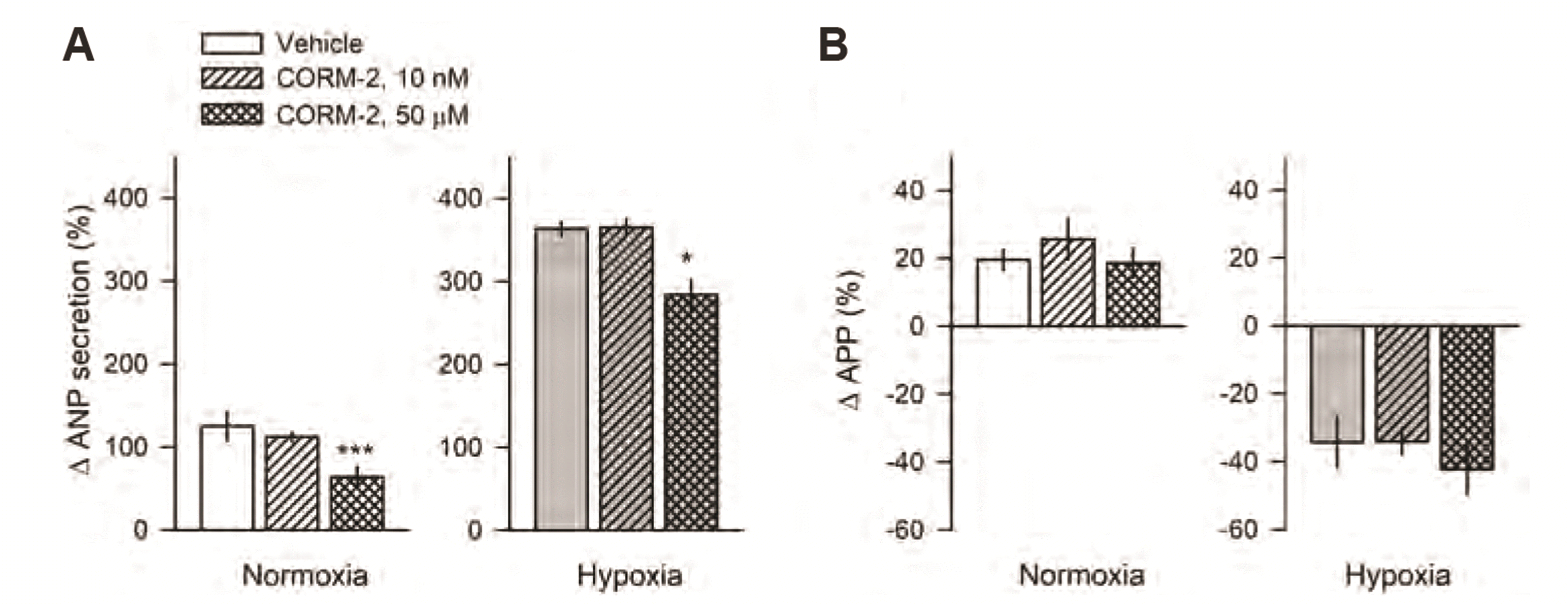
Modulation of ANP secretion induced by CORM-2 during hypoxic condition
Low (10 nM) and high doses (50 µM) of CORM-2 were used to compare the effect of CORM-2 on ANP secretion in normoxic and hypoxic conditions. In normoxic conditions, a high dose of CORM-2 suppressed ANP secretion (63.8 ± 12.1% vs. 124.9 ± 17.6%) (Fig. 5A), but a low dose did not. In hypoxic conditions, change in high stretched-ANP secretion was significantly higher (363.7 ± 8.5% vs. 124.9 ± 17.6%, p < 0.01) (Fig. 5A) and change in APP was lower (–34.3 ± 7.5% vs. 19.6 ± 2.9%, p < 0.01) than in normoxic conditions (Fig. 5B). During hypoxia, CORM-2 50 µM suppressed ANP secretion (283.7 ± 18.4% vs. 363.7 ± 8.5%, p < 0.05; Fig. 4A). CORM-2 suppressed ANP secretion in both normoxic (by approximately 50%) and hypoxic conditions, (by approximately 20%). No significant differences in APP were observed after administration of CORM-2 (Fig. 5B).
DISCUSSION
Our experiment showed that high doses of CORM-2 and hemin suppressed high stretch-induced ANP secretion, while CORM-3 and ZnPP did not suppress ANP secretion. The order of potency for the suppression of ANP secretion was hemin > CORM-2 >> CORM-3. The suppression of ANP secretion by CORM-2 was blocked by pretreatment with 5-HD, paxilline, and ODQ, but not by pretreatment with diltiazm, wortmannin, LY, and L-NAME. The effect of CORM-2 on ANP secretion appeared to have been attenuated in hypoxic conditions. These results suggest that CORM-2 suppresses ANP secretion via a mitochondrial KATP channel, a BKCa channel, and sGC pathway.
The beneficial effects of gasotransmitters generally manifest with lower doses, while the effects diminish at higher doses. In some cases, a higher dose may result in harmful levels of toxicity. Several recent studies have suggested that administration of a low level of CO either by CO inhalation or CORMs can protect the heart, liver, and kidneys against I/R [12,32,33]. In addition, low concentrations of CO also exhibit pleiotropic biological functions, including anti-apoptotic, cytoprotective, and anti-inflammatory effects [11-13]. We hypothesized that these protective effects may partly relate to the cardiac hormone ANP. Our aim, therefore, was to determine any direct effect of CO on ANP secretion, as well as the associated atrial dynamics, in normal isolated rat atria. Our experiments employed CORM-2 and HO activator (hemin) across a wide range of concentrations. At low concentrations (between 0.1 and 100 nM) CORM-2 and hemin had no significant effect on ANP secretion. At higher doses, stretch-activated ANP secretion was suppressed by CORM-2 and hemin. The dose-response curve of CORM-2 revealed no significant change in ANP secretion prior to 10 µM concentration. ANP secretion levels thereafter abruptly decreased with the administration of 50 µM and 100 µM of CORM-2. The dose-response curve of hemin was very similar to that of CORM-2, excepting a notable difference in potency. Our results are in broad agreement with a previous report showing that a characteristic gaseotransmitter pharmacological response assumes the form of a bell-shaped dose-response curve [4,26]. A number of prior studies testing the beneficial effects of the administration of endogenous and exogenous CO in the case of I/R injury have been performed, with rat, mouse, and cardiac cell cultures exposed variously to CO [32], CORM-2 [33], and CORM-3 [12,34]. We compared the suppressive effect of CORM-2 on ANP secretion to that of CORM-3 and HO inhibitor. Notably, neither CORM-3 nor HO inhibitor (ZnPP) showed any significant effect on ANP secretion. While we are unable to explain why the effect of CORM-3 on ANP secretion is different from that of CORM-2, we hypothesize that it may be due to different rates of CO release.
CO can cause vaso-relaxation by activating sGC, increasing cGMP, and activating the BKCa channel [26,35-37]. K+ channel activation leads to membrane hyperpolarization followed by an inhibition of calcium entry via voltage operated Ca2+ channels. However, both cGMP-dependent and -independent signaling mechanisms are involved in the vascular action of CO [4,26]. It has also been reported that CORM-2-mediated cardioprotection may be a result of the activation of the p38 MAPK and protein kinase C pathways prior to ischemia, and the PI3K pathway during reperfusion [33]. Therefore, to elucidate the underlying signaling mechanisms involved in CORM-2-induced suppression of ANP secretion, atria were pretreated with several modulators. The suppression of ANP secretion by CORM-2 was completely blocked by pretreatment with the BKCa channel inhibitor (paxilline) and the mitochondrial KATP channel blocker (5-HD), and partially reduced by the sCG inhibitor. These results are consistent with a prior report showing complete blockage of a CORM-2 induced electrical current by a BKCa channel inhibitor [37] and a decrease in vascular resistance by CO through sGC activation and Kca channel opening in a mouse kidney [38]. However, diltiazem and wortmannin (including LY) did not block the effects of CORM-2. Another previous study has demonstrated that, with respect to CORM-2 mediated cardioprotection, PI3K is important during reperfusion rather than ischemia [33]. It follows that the differences in the signaling pathway of CORM-2 may be due to exposure time and duration to modulator and experimental tissues.
We also compared the effect of low and high doses of CORM-2 on ANP secretion in hypoxic conditions, which HO-1 can be induced. In the normoxic condition, CORM-2 (50 µM) suppressed ANP secretion by 50% compared to the control group (65% vs. 125%) whereas in a hypoxic condition, CORM-2 suppressed ANP secretion by 20% (280% vs. 360%). No change in the expression of HO-1 protein was observed in atria exposed to a hypoxic condition (data not shown). The attenuation of CORM-2’s suppressive effect may be due to modulation of the KATP and BKCa channels during hypoxia.
In summary, these results suggest that CORM-2 suppresses ANP secretion via the mitochondrial KATP channel, BKCa channel, and sGC pathway.




 XML Download
XML Download