

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Coronary Atherosclerosis (AS) is a multifactorial chronic disease characterized by accumulation of lipids and fibrous components in the aorta [1,2]. In the aortic tunica media, vascular smooth muscle cells (VSMCs) are the main cell type [3,4], and the proliferation and migration of VSMCs in intima are associated with the formation of atherosclerotic plaques [5,6]. Due to the long clinical treatment cycle, poor prognosis, and frequent relapse of coronary AS, it is urgent to explore a novel therapeutic target for Coronary AS from the molecular level.
MicroRNAs (miRNAs) are small non-coding RNA molecules that can regulate gene expression at the post-transcriptional level [7-9]. MiRNAs can directly or indirectly modulate plaque formation, which involves the regulation of VCMCs, so that to affect the pathophysiology of coronary AS and cardiovascular disease [8,10]. Numerous miRNAs have been reported to participate in functionally regulating VSMCs during AS. For examples, miR-499a-3p and miR-135b-5p promote AS development by targeting MEF2C [11]. Overexpression of miR-448 promotes VSMC proliferation and migration via MEF2C [12]. MiR-365 participates in coronary AS through regulating IL-6 expression [13]. MiR-491-5p is abnormally expressed in cancers [14-16]. Notably, a previous study revealed that miR-491-5p is lowly expressed in the atherosclerotic plaque tissues and plasma samples of AS patients, and inhibited the growth and migration of VSMCs by targeting MMP-9 [17], suggesting its meaningful role in the AS development. Moreover, miR-491-5p also increases the risk of atherosclerotic cerebral infarction through modulation of MMP-9 gene, as reported in another study [18].
In this study, we examined the expression of ten potential mRNAs in plaque tissues and serum from coronary AS patients and identified miR-491-5p as an upstream regulator. We hypothesized that miR-491-5p regulated the dysfunctions of VSMCs, which may contribute to novel research strategies for the coronary AS.
Go to : 
METHODS
Clinical specimens
This studied included 22 coronary AS patients (16 males and 6 females; 9 cases aged ≤ 50; 13 cases aged > 50) with coronary endarterectomy at our hospital. Clinical characteristics of patients with coronary AS were provided in Table 1. The healthy control group included 13 patients (6 males and 7 females; 5 cases aged ≤ 50; 8 cases aged > 50) who underwent physical examinations in the same period at our hospital. The patients with complications or infections of liver or kidney, diabetes, or tumors were excluded. The Gensini scoring system was applied to evaluate the degree of AS. Written informed consent was obtained from all participants. The study complied with the Declaration of Helsinki and was approved by the Ethics Committee of The Affiliated Huaian No. 1 People’s Hospital of Nanjing Medical University (IRB: 2016-045, Jiangsu, China).
Plaque tissues and adjacent intimal tissues were taken by coronary endarterectomy and then stored in liquid nitrogen. Peripheral blood was collected, and serum was separated by centrifugation at 400 g for 10 min.
Cell culture and transfection
VSMCs were dissected from perioperative aortic punch samples of participating patients as previous study described [19]. The dissected VSMCs were then cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% Gibco fetal bovine serum (FBS; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 100 µg/ml penicillin-streptomycin (Sigma-Aldrich) at 37°C and 5% CO2. The 10 nM miR-491-5p mimics were used for miR-491-5p overexpression and an equal quantity of miR-491-5p antagomir was used for miR-370 downregulation. Simultaneously, NC mimics and NC antagomir was used in the present study. To downregulate UBE2G2, SLC16A3, and PNO1 expression, short hairpin RNAs targeting UBE2G2, SLC16A3, and PNO1 (sh-UBE2G2, sh-SLC16A3, and sh-PNO1) were used. The miR-491-5p mimic, miR-491-5p antagomir, sh-UBE2G2, sh-SLC16A3, sh-PNO1, and their relevant controls were all synthesized by GenePharma (Shanghai, China). Cell transfections were conducted for 48 h using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Reverse transcription quantitative polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen). Synthetic spike-in miRNAs (MiRXES, Singapore) was prepared for miRNAs to obtain RNA samples, which was then reverse transcribed into cDNA. Reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis was performed using mSMRT-qPCR miRNA assay platform (MiRXES, Singapore). The relative expression levels of miRNAs were normalized to U6, and the relative expression levels of mRNAs were normalized to GAPDH, which were all quantified using the 2−ΔΔCq method. Sequences of primers are provided in Table 2.
Table 2
Primers used for quantitative RT-PCR
![]()
Western blot analysis
Total protein extraction was performed, and the protein concentration was measured. Proteins (25 µg) were then loaded on 10% SDS-PAGE and transferred onto PVDF membranes. After blocking with non-fat milk (5%) at room temperature for 1 h, the membranes were then incubated with the primary antibodies below at 4°C overnight: UBE2G2 (ab174296; 1:1,000; Abcam, Shanghai, China), SLC16A3 (ab244385; 1:1,000; Abcam), POLR2C (ab182150; 1:100; Abcam), PNO1 (ab227978; 1:1,000; Abcam), AMDHD2 (ab133306; 1:1,000; Abcam), α-SMA (ab5831; 1:1,000; Abcam), calponin (ab46794; 1:5,000; Abcam), SM22α (ab14106; 1:1,000; Abcam), smoothelin (ab8969; 1:100; Abcam), GAPDH (as a loading control; ab8245; 1:500; Abcam). Subsequently, the secondary antibody of HRP-conjugated goat anti-rabbit IgG (1:3,000) was added and incubated at room temperature for 1 h. Finally, the protein bands were detected by the ECL Plus Kit (Pierce, Rockford, IL, USA), The relative protein expression was analyzed using ImageJ software 1.4.
RNA pull down assay
Bio miR-491-5p and Bio NC were co‐transfected to VSMCs for 2 days. VSMCs (1 × 107) were dissolved in the soft lysis buffer plus RNasin (80 U/ml; Promega, Madison, WI, USA). Subsequently, streptavidin agarose beads (Life Technologies, Rockville, MD, USA) were incubated with the reaction mixture for 1 h at room temperature. RT-qPCR was prepared to evaluate the co-precipitated RNAs.
Cell counting kit-8 assay
VSMCs were seeded in 96-well plates (5 × 103 cells/well) for 24 h at 37°C and then used to assess cell proliferation. After the transfection, the cell proliferation value was measured by a cell counting kit 8 (CCK-8 kit; Thermo Fisher Scientific, Inc.) at 24, 48, and 72 h. CCK-8 reagent (10 µl) was then added and incubated at 37°C for 1 h. Subsequently, the optical density was measured at 450 nm.
Wound healing assay
VSMCs were cultured in 6-well plates and cultured for 24 h. Subsequently, the parallel lines were scratched on the cell culture plate with a 200 µl pipette tip. Following washing with PBS three times, cells were cultured in serum-free DMEM for an additional 48 h. The wounded gaps were photographed at different time points (0 and 24 h).
Statistical analyses
Data are shown as the mean ± standard deviation. The Student’s t-test was used to assess the differences between two groups. Two-way ANOVA was conducted for difference comparison in groups with two independent variables. All data were statistically analyzed by the SPSS 21.0 (IBM Co., Armonk, NY, USA) statistical software. p-values of less than 0.05 was defined as significant.
Go to :
RESULTS
Expression of 10 mRNAs in plaques and adjacent intimal tissues from AS patients
According to the gene profiling by array results of GSE132651 which determined differences in blood outgrowth endothelial cells from subjects with normal or abnormal coronary endothelial function, top ten genes were differentially expressed (Table 3). Subsequently, we measured the expression levels of UBE2G2, SNORD45C, SNORD45A, SNORD45B, RABGGTB, SLC16A3, POLR2C, PNO1, CEMP1, AMDHD2 using RT-qPCR. The levels of UBE2G2, SNORD45C, SNORD45A, SLC16A3, POLR2C, PNO1, AMDHD2 in plaques from coronary AS patients were significantly higher than those in adjacent intima. On the contrary, CEMP1 expression was downregulated in plaques from coronary AS patients, compared with that in adjacent intima. However, the expression levels of SNORD45B and RABGGTB had no significant difference in plaques and adjacent intima from coronary AS patients (Fig. 1A). Only mRNAs (UBE2G2, SNORD45C, SNORD45A, SLC16A3, POLR2C, PNO1, CEMP1, AMDHD2) with significant difference were listed in Fig. 1B.
 | Fig. 1Expression of 10 mRNAs in plaques and adjacent intimal tissues from atherosclerosis (AS) patients.(A) Expression levels of ten mRNAs (UBE2G2, SNORD45C, SNORD45A, SNORD45B, RABGGTB, SLC16A3, POLR2C, PNO1, CEMP1, AMDHD2) in plaques and adjacent intima from coronary AS patients were measured using RT-qPCR with pared t-test, as presented by the scatter diagram. (B) mRNAs (UBE2G2, SNORD45C, SNORD45A, SLC16A3, POLR2C, PNO1, CEMP1, AMDHD2) with significant difference were listed in the bar graph. *p < 0.05, **p < 0.01, NS indicates no significance.
|
Table 3
Top 10 genes that are differentially expressed in blood outgrowth endothelial cells from subjects with normal or abnormal coronary endothelial function
![]()
Expression of 10 mRNAs in serum from AS patients and healthy controls
The levels of UBE2G2, SNORD45B, RABGGTB, SLC16A3, POLR2C, PNO1, and AMDHD2 in the serum of coronary AS patients were significantly higher than those in the serums of healthy patients. While the expression levels of SNORD45C, SNORD45A, and CEMP1 had no significant difference in the serum of coronary AS patients and healthy patients (Fig. 2A). In Fig. 2B, mRNAs (UBE2G2, SNORD45B, RABGGTB, SLC16A3, POLR2C, PNO1, and AMDHD2) with significant difference were listed.
 | Fig. 2Expression of 10 mRNAs in serum from atherosclerosis (AS) patients and healthy controls.(A) Expression levels of ten mRNAs (UBE2G2, SNORD45C, SNORD45A, SNORD45B, RABGGTB, SLC16A3, POLR2C, PNO1, CEMP1, AMDHD2) in the serum of coronary AS patients and healthy patients were measured using RT-qPCR with unpaired t-test. (B) mRNAs (UBE2G2, SNORD45C, SNORD45A, SLC16A3, POLR2C, PNO1, CEMP1, AMDHD2) with significant difference were listed. *p < 0.05, **p < 0.01, ***p < 0.001, NS indicates no significance.
|
Potential upstream miRNAs for the abnormally expressed mRNAs
We found that five mRNAs (UBE2G2, SLC16A3, POLR2C, PNO1, and AMDHD2) were significantly abnormally expressed in both plaques and serums from subjects with coronary AS. TargetScan was used to predict the potential upstream miRNAs of these five genes, and 11 common miRNAs (hsa-miR-491-5p, hsa-miR-4645-5p, hsa-miR-4673, hsa-miR-6791-5p, hsa-miR-6727-3p, hsa-miR-6747-3p, hsa-miR-4308, hsa-miR-4722-3p, hsa-miR-4292, hsa-miR-6852-5p, and hsa-miR-7160-5p) were identified (Fig. 3A). We them examined the expression levels of these miRNAs using RT-qPCR. Only miR-491-5p showed significantly downregulated expression in plaques from coronary AS patients, compared with that in adjacent intima (Fig. 3B). MiR-491-5p expression was also decreased in the serum of coronary AS patients, compared with that in the serums of healthy patients (Fig. 3C). Collectively, miR-491-5p was a potential upstream miRNA for abnormally expressed UBE2G2, SLC16A3, POLR2C, PNO1, and AMDHD2.
 | Fig. 3Potential upstream miRNAs for the abnormally expressed mRNAs.(A) TargetScan predicted the potential upstream miRNAs of UBE2G2, SLC16A3, POLR2C, PNO1, and AMDHD2. (B) Expression levels of 11 miRNAs in plaques and adjacent intima from coronary AS patients were examined using RT-qPCR with pared t-test. (C) Expression levels of 11 miRNAs in the serum of coronary atherosclerosis (AS) patients and healthy patients were examined using RT-qPCR with unpaired t-test. *p < 0.05, **p < 0.01.
|
MiR-491-5p negatively regulates UBE2G2, SLC16A3, PNO1 expression in VSMCs
To confirm the predicted regulatory role of miR-491-5p on target genes, we conducted the subsequent experiments in VSMCs separated from the aortic wall sections of coronary AS patients. According to the results of RT-qPCR, miR-491-5p expression was higher in the VSMCs transfected with miR-491-5p mimics than that in the control group (Fig. 4A). mRNA expression and protein levels of UBE2G2, SLC16A3 and PNO1 were significant downregulated in the cells transfected with miR-491-5p mimics, compared with those in the control group, as evaluated by RT-qPCR and western blotting, respectively (Fig. 4B, C). However, the mRNA expression and protein levels of POLR2C and AMDHD2 had no significant difference in the cells transfected with miR-491-5p mimics and NC mimics. Similarly, RNA pull down assay revealed that the expression levels of UBE2G2, SLC16A3 and PNO1 were upregulated in VSMCs transfected with Bio miR-491-5p, compared with those in control groups (Fig. 4D). These results indicated that the expression levels of UBE2G2, SLC16A3 and PNO1 were negatively regulated by miR-491-5p.
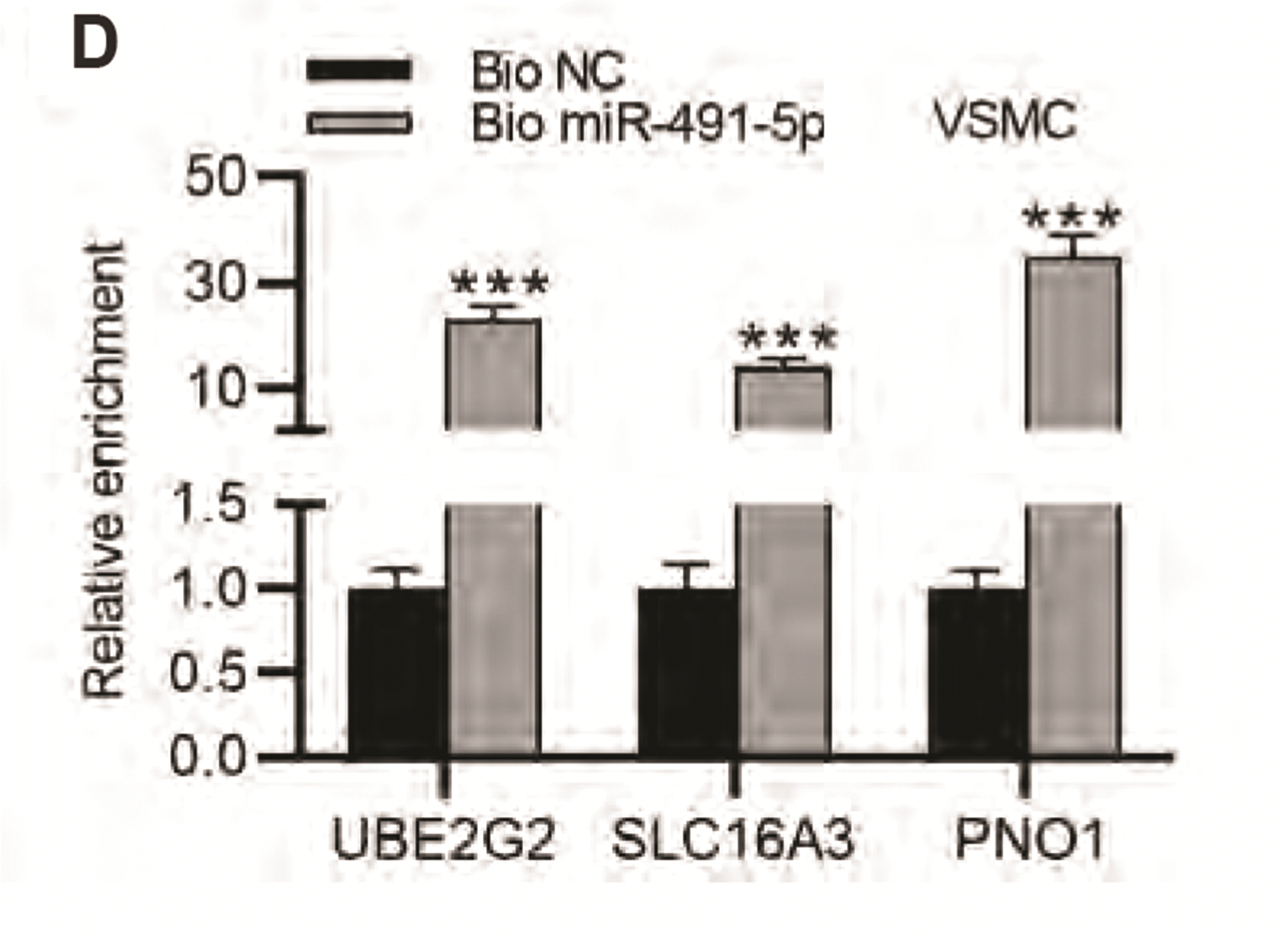 | Fig. 4MiR-491-5p negatively regulates UBE2G2, SLC16A3, PNO1 expression in vascular smooth muscle cells (VSMCs).(A) Overexpression efficiency of miR-491-5p in VSMCs was evaluated using RT-qPCR. (B) Expression levels of UBE2G2, SLC16A3, POLR2C, PNO1, and AMDHD2 in VSMCs transfected with miR-491-5p mimics and NC mimics were examined by RT-qPCR. (C) Protein levels of UBE2G2, SLC16A3, POLR2C, PNO1, and AMDHD2 in VSMCs transfected with miR-491-5p mimics and NC mimics were examined by Western blot. (D) RNA pull down assay was for detecting whether UBE2G2, SLC16A3, and PNO1 bind with miR-491-5p. *p < 0.05, **p < 0.01, ***p < 0.001.
|
MiR-491-5p inhibits viability, migration, and increased expression of contractile markers in VSMCs
The function of miR-491-5p in VSMCs was then explored. CCK-8 revealed that the viability was suppressed in VSMCs transfected with miR-491-5p mimics, compared with that in control group (Fig. 5A). According to the results of the wound healing assay, the migration capacity was decreased in VSMCs transfected with miR-491-5p mimics, compared with that in control group (Fig. 5B). Importantly, the protein levels of contractile markers in VSMCs were elevated by miR-491-5p mimics (Fig. 5C). These results suggested that miR-491-5p inhibited viability, migration, and increased expression of contractile markers in VSMCs.
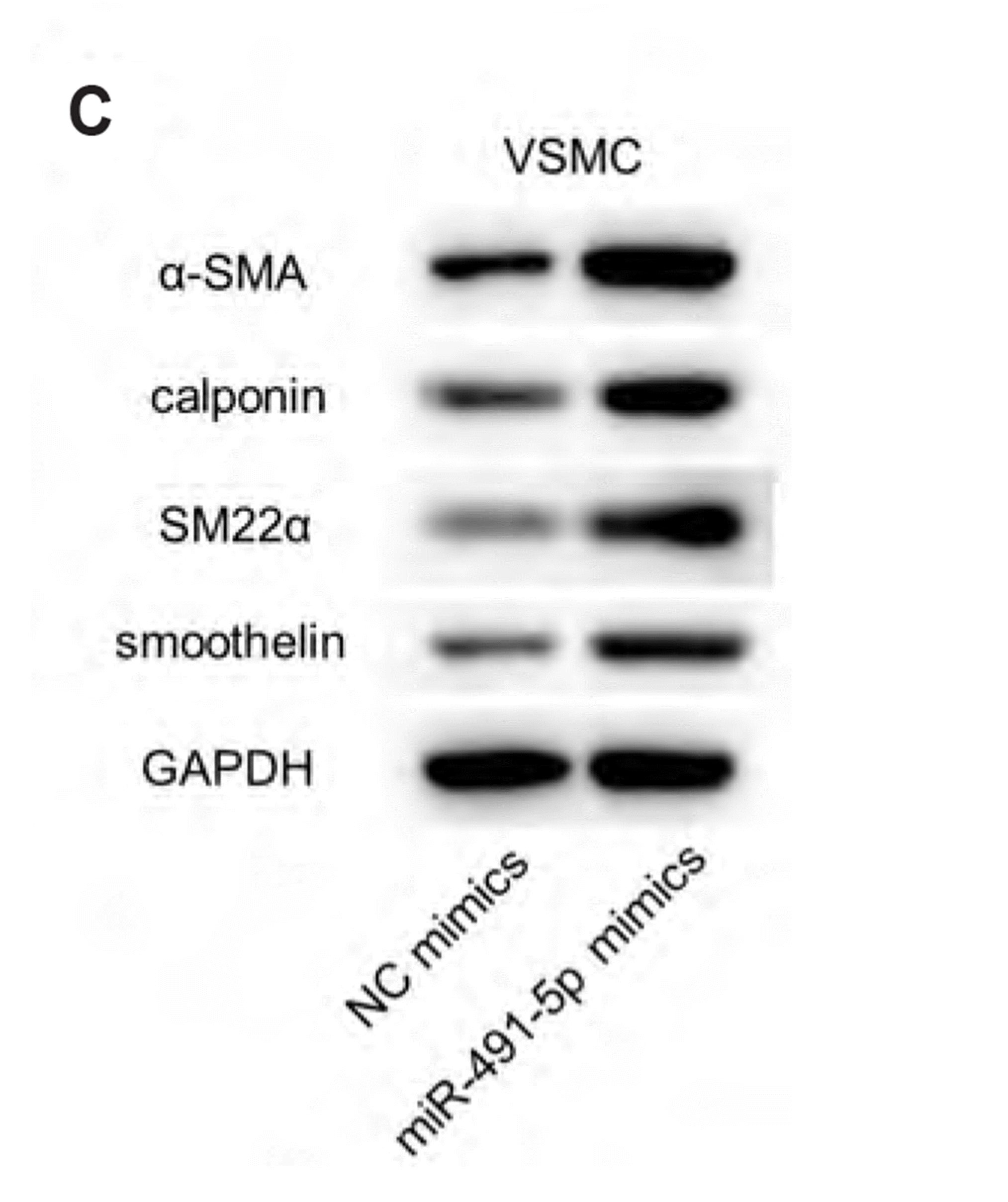 | Fig. 5MiR-491-5p inhibited viability, migration, and increased expression of contractile markers in vascular smooth muscle cells (VSMCs).(A) Cell counting kit 8 (CCK-8) revealed the viability in VSMCs with transfection of miR-491-5p mimics and NC mimics. (B) Wound healing was performed to detect the migration capacity of VSMCs with transfection of miR-491-5p mimics and NC mimics. (C) The protein levels of contractile markers (α-SMA, calponin, SM22α, and smoothelin) in VSMCs with transfection of miR-491-5p mimics and NC mimics were measured by Western blot. *p < 0.05, **p < 0.01.
|
MiR-491-5p antagomir promotes viability, migration, and increased expression of contractile markers in VSMCs
To further confirm the function of miR-491-5p and its target genes UBE2G2, SLC16A3, and PNO1 in VSMCs, miR-491-5p antagomir, sh-UBE2G2, sh-SLC16A3, and sh-PNO1 were transfected in VSMCs and NC antagomir and sh-NC were used as negative control. A CCK-8 assay revealed that silencing miR-491-5p in VSMCs significantly promoted cell proliferation when compared with the control group at 24, 48, and 72 h, while UBE2G2, SLC16A3 and PNO1 knockdown partially reversed the changes (Fig. 6A). Wound healing assay was performed to assess cell migration. The results indicated that the inhibition of miR-491-5p significantly upregulated VSMCs migration. However, the upregulation was then alleviated by UBE2G2, SLC16A3, and PNO1 knockdown (Fig. 6B). Moreover, the protein levels of α-SMA, calponin, SM22α, and smoothelin were suppressed by miR-491-5p antagomir, While UBE2G2, SLC16A3, and PNO1 knockdown partially restored the changes, as shown by western blotting (Fig. 6C). Taken together, these results suggested that miR-491-5p silencing promotes VSMCs proliferation and migration but inhibits the expression of contractile markers. However, UBE2G2, SLC16A3, and PNO1 knockdown reversed the changes induced by miR-491-5p silencing.
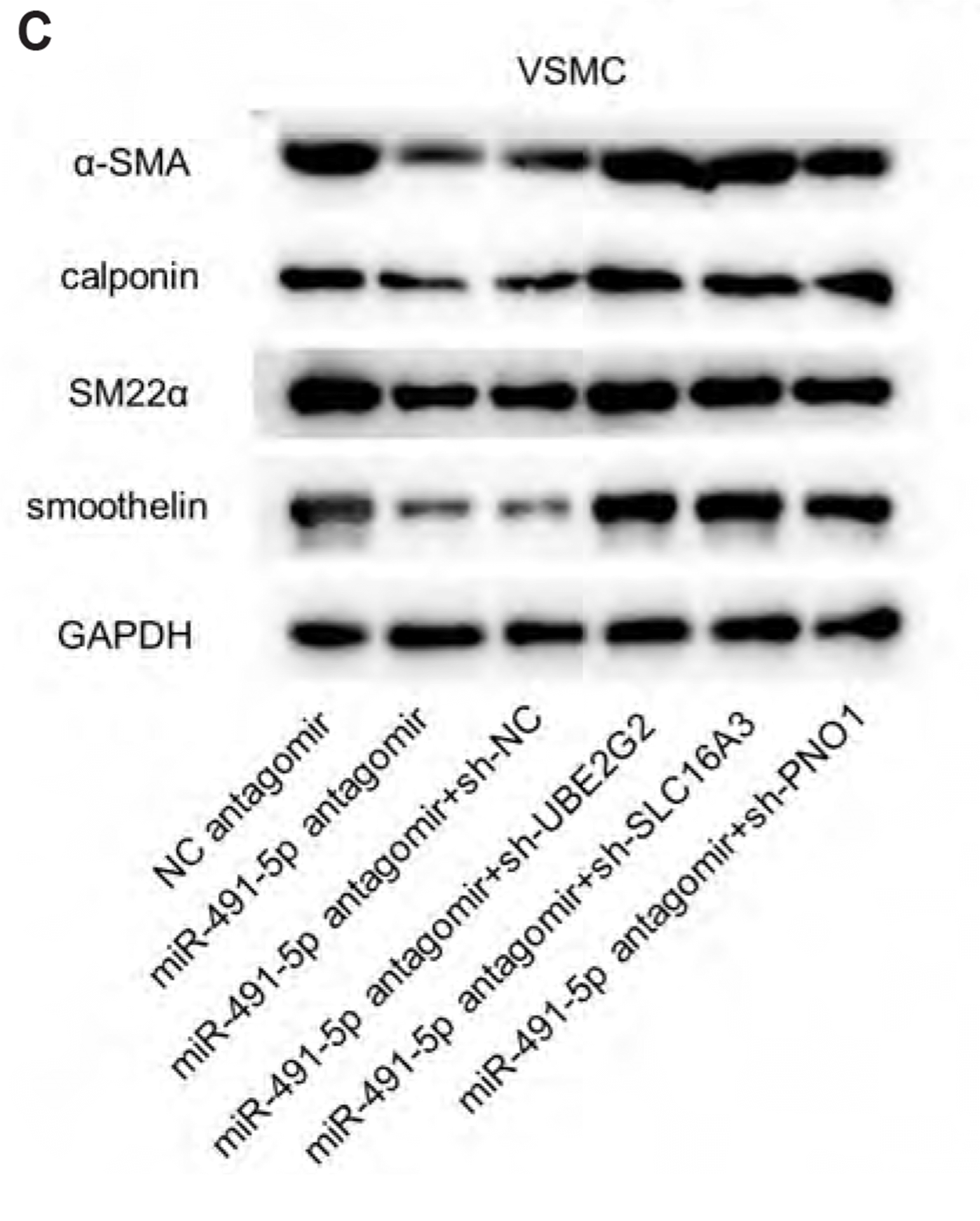 | Fig. 6MiR-491-5p antagomir promotes viability, migration, and increased expression of contractile markers in vascular smooth muscle cells (VSMCs).(A) CCK-8 revealed the viability in VSMCs with transfection of NC antagomir, miR-491-5p antagomir, miR-491-5p antagomir+sh-NC, miR-491-5p antagomir+sh-UBE2G2, miR-491-5p antagomir+sh-SLC16A3, and miR-491-5p antagomir+sh PNO1. (B) Wound healing was performed to detect the migration capacity of VSMCs with transfection of NC antagomir, miR-491-5p antagomir, miR-491-5p antagomir+sh-NC, miR-491-5p antagomir+sh-UBE2G2, miR-491-5p antagomir+sh- SLC16A3, and miR-491-5p antagomir+sh PNO1. (C) The protein levels of contractile markers (α-SMA, calponin, SM22α, and smoothelin) in VSMCs with transfection of NC antagomir, miR-491-5p antagomir, miR-491-5p antagomir+sh-NC, miR-491-5p antagomir+sh-UBE2G2, miR-491-5p antagomir+sh- SLC16A3, and miR-491-5p antagomir+sh PNO1. *p < 0.05, **p < 0.01.
|
Go to :
DISCUSSION
In our study, we first confirmed ten potential mRNAs that were differentially expressed in blood outgrowth endothelial cells from subjects with normal or abnormal coronary endothelial function, according to the gene profiling by array results of GSE132651. After evaluating their expression levels, we found that expression of five mRNAs (UBE2G2, SLC16A3, POLR2C, PNO1, and AMDHD2) had significant difference in both plaques and serum from coronary AS patients, compared with those in adjacent intimal tissues from coronary AS patients or serum from healthy controls. TargetScan then predicted 11 common upstream miRNAs of these mRNAs. Notably, only miR-491-5p showed significantly downregulated expression levels in plaques and serum from coronary AS patients, compared with those in adjacent intimal tissues from coronary AS patients or serum from healthy controls.
MiRNAs are short (approximately 24 nucleotides), noncoding RNAs that bind to the 3’ untranslated region of targeted mRNAs and induce degradation or translational inhibition [20]. To date, numerous studies have shown that miRNAs are effective regulators of VSMC differentiation [21-24]. These miRNAs regulate the VSMC differentiation and coronary AS mainly through the modulation of target gene expression [3,25,26]. Previous studies have reported several miRNA-mRNA regulatory networks in AS [27-29]. For instance, upregulated miR-93 contributes to coronary AS pathogenesis through targeting ABCA1 [30]. MiR-126 represses the progression of coronary AS in the mice by binding to S1PR2 [31]. MiR-491-5p has been once explored in VSMCs in a previous study, which demonstrates its inhibitory role on the growth and migration of VSMCs by binding with MMP-9 gene, indicating the involvement of miR-491-5p in AS development [17]. Particularly, we found three mRNAs UBE2G2, SLC16A3, and PNO1 that were negatively regulated by miR-491-5p, which were not mentioned in any previous research. Mechanically, UBE2G2, SLC16A3, and PNO1 were validated as the targets downstream miR-491-5p, indicating that the upregulation of UBE2G2, SLC16A3, and PNO1 in plaques and serum from AS patients was induced by the downregulation of miR-491-5p. Furthermore, the functions of miR-491-5p, UBE2G2, SLC16A3, and PNO1 in smooth muscle cells were assessed. Notably, we found that miR-491-5p overexpression inhibited viability, migration, and increased the expression of contractile markers α-SMA, calponin, SM22α, and smoothelin in VSMCs. The increase of these contraction markers suggests that miR-491-5p overexpression promoted the differentiation of phenotypic proteins, which is a key element in AS development because the synthetic phenotype of VSMCs can migrate, proliferate, and generate extracellular matrix proteins [5]. Inversely, silencing miR-491-5p significantly promoted VSMCs viability and migration, and inhibited the expression of α-SMA, calponin, SM22α, and smoothelin in VSMCs. However, UBE2G2, SLC16A3, and PNO1 knockdown reversed the changes induced by miR-491-5p antagomir.
In conclusion, this study revealed the differentially expressed mRNAs and their upstream miR-491-5p in patients with coronary atherosclerosis. We also found that miR-491-5p inhibited viability, migration, and increased expression of contractile markers via regulating target genes UBE2G2, SLC16A3, and PNO1 in VSMCs. These results might provide a novel insight into the molecular mechanisms underlying the miRNA-controlled VSMCs dysfunctions in diagnostic and therapeutic approaches against coronary AS.
Go to :
 XML Download
XML Download