

 PDF
PDF Citation
Citation Print
Print



Introduction
Lung cancer is the second most common cancer world-wide and the leading cause of cancer-related deaths [1]. Non–small cell lung cancer (NSCLC) accounts for approximately 85% of all new lung cancer cases and more than two-thirds of these cases are metastatic at the time of diagnosis [2]. For decades, cytotoxic chemotherapy in the form of a platinum-doublet was the standard of care for advanced stage disease. While these therapies are associated with increased survival and improved quality of life, the responses achieved by chemotherapeutic agents are modest and there is a lack of durable clinical benefit [3–5].
Advances in genomics leading to identification of oncogenic driver gene aberrations have revolutionized the practice of thoracic oncology. Significant improvements in response rates and greater than 3-year median survival in the metastatic setting is now seen in subsets of patients. Although molecular targeted therapies were initially used in the late 1990s in patients with NSCLC in the form of gefitinib, an oral epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI), initial response rates were approximately 10% for the unselected NSCLC population. IPASS (Iressa Pan-Asia Study) was the first phase III study which demonstrated an improved outcome with oral gefitinib when compared with carboplatin-paclitaxel in patients with EGFR-mutant NSCLC [6]. In this trial, the objective response rate with oral gefitinib was 71% versus 1% between the EGFR-mutant and wild-type patients respectively [6]. Since then, multiple other TKIs targeting specific molecular pathways have been developed. Targeted therapies inhibit protein products of aberrant genes that drive tumors to continuously proliferate and survive. Drugs have been developed with a wide range of mechanisms of action. One of the most common drug mechanisms is the inhibition of tyrosine phosphorylation of target proteins by small molecule TKIs that compete with ATP-binding to the tyrosine kinase domain of proteins and thereby block protein phosphorylation and inhibit the proliferation and oncogenic signals through a number of signaling pathways such as the RAS-MAPK (mitogen-activated protein kinase), PI3K (phosphoinositide 3-kinase)-AKT-mTOR (mammalian target of rapamycin), and JAK/STAT pathways [7]. Drugs like osimertinib, alectinib, larotrectinib, crizotinib, among others are in this class. In contrast, the MEK inhibitor trametinib is an ATP non-competitive inhibitor that binds MEK adjacent to the ATP-binding site in common with other MEK allosteric inhibitors [8]. A different mechanism is also exhibited by the KRAS G12C inhibitors such as sotorasib, which binds covalently to the KRAS mutated cysteine 12 residue at the switch II pocket [9]. Emerging evidence has demonstrated that antibody-drug conjugates (ADCs) are active in NSCLC. In essence, ADCs are composed of a monoclonal antibody linked to a cytotoxic payload via a linker. They are designed to deliver these drugs to cells that express the target antigen of the selected antibody [10]. As an example, trastuzumab deruxtecan is an ADC which consists of a humanized immunoglobulin G1 antibody against human epidermal growth factor receptor 2 (HER2) conjugated to a topoisomerase I inhibitor payload by a cleavable tetrapeptide-based linker. Recent clinical data demonstrate encouraging results in NSCLC [11,12]. Currently, oncogenic aberrations in eight genes (EGFR, ALK, ROS1, BRAF, KRAS, NTRK, MET, and RET) have U.S. Food and Drug Administration (FDA) approved therapies. These agents have typically been used in the metastatic setting (Table 1) [13–35]. However, the use of these agents in earlier stages of disease have gained recent interest. A phase III study of osimertinib in early-stage resectable EGFR-mutant NSCLC has shown improvement in disease-free survival, and now is FDA approved [36]. Various clinical trials designed to incorporate other targeted therapies in the adjuvant setting are underway [37]. As we attempt to enhance the excellent clinical activity of these agents and develop curative strategies, complexities and challenges emerge. This review discusses some of the challenges in the use of targeted therapies in NSCLC and provides guidance for future strategies. The major challenges hampering the expanded use of targeted agents to potentially achieve cure of advanced NSCLC are resistance in its different forms, toxicity, and high cost of these agents limiting access by all NSCLC patients.
Drug Resistance
There are varied responses of NSCLC patients’ tumors to targeted therapies. A few patients will achieve a complete response, others a partial response and yet in some others, there is no objective response to therapy. Even with our most effective agents, the best initial response rates are around 70%–80% [18,38]. The tumors that fail to respond are deemed to have primary resistance. Unfortunately, even in those who do respond, disease progression inevitably occurs due to acquired resistance. These resistance mechanisms, primary and acquired resistance, are discussed below.
1. Primary resistance
Primary resistance or intrinsic resistance can be defined as the lack of initial treatment response despite the presence of a targetable driver mutation. Although there is a paucity of data, various factors are known to play a role in primary resistance. Differential sensitivities to TKI therapy based on mutation-subtype, pharmacology of the drug and adherence to treatment play a significant role [39–42]. Tumor cells within the same tumor site or between different metastatic tumor sites within the same host do not necessarily share uniform characteristics, including histology, metastatic or proliferative potential and molecular signatures. This is referred to as tumor heterogeneity [43]. At the histologic level, one can have adeno-squamous cancers or pleomorphic cancer with adenocarcinoma or adenocarcinoma features. Additionally, many studies have shown significant genetic diversity in the same patients from different metastatic sites [41,42]. Jamal-Hanjani et al. [41], in their prospective study, performed multi-region whole-exome sequencing in 100 patients at 327 tumor regions with early-stage NSCLC after surgical resection but before the receipt of adjuvant chemotherapy. The study demonstrated extensive intratumor heterogeneity suggesting genomic instability processes during tumor development. Although originating from the same clone, it has been hypothesized that the tumor cells adapt to various environmental insults via a Darwinian-like selection mechanism and a subpopulation of cells undergo genotypic and/or phenotypic changes to transform themselves to drug-resistant clones [44]. Tumor heterogeneity therefore plays a crucial role in disease progression especially in patients with an oncogenic-addicted lung cancer where a TKI is utilized to target a specific driver mutation [45].
2. Acquired resistance
There is presumed acquired resistance when there is tumor progression after an initial objective response. In 2010, Jackman et al. [46] provided a guideline to define acquired resistance to EGFR TKIs in NSCLC. These general concepts, with some adaptations, can be used as a general guide for the clinical definition of acquired resistance. In summary, for acquired resistance all patients should have the following criteria: Previous treatment with a targetable drug; either or both of the following: a tumor that harbors an aberration known to be associated with drug sensitivity or objective clinical benefit from treatment with a targetable drug; systemic progression of disease following Response Evaluation Criteria in Solid Tumor or World Health Organization criteria while on continuous treatment and no intervening systemic therapy between cessation of targetable drug and initiation of new therapy [46].
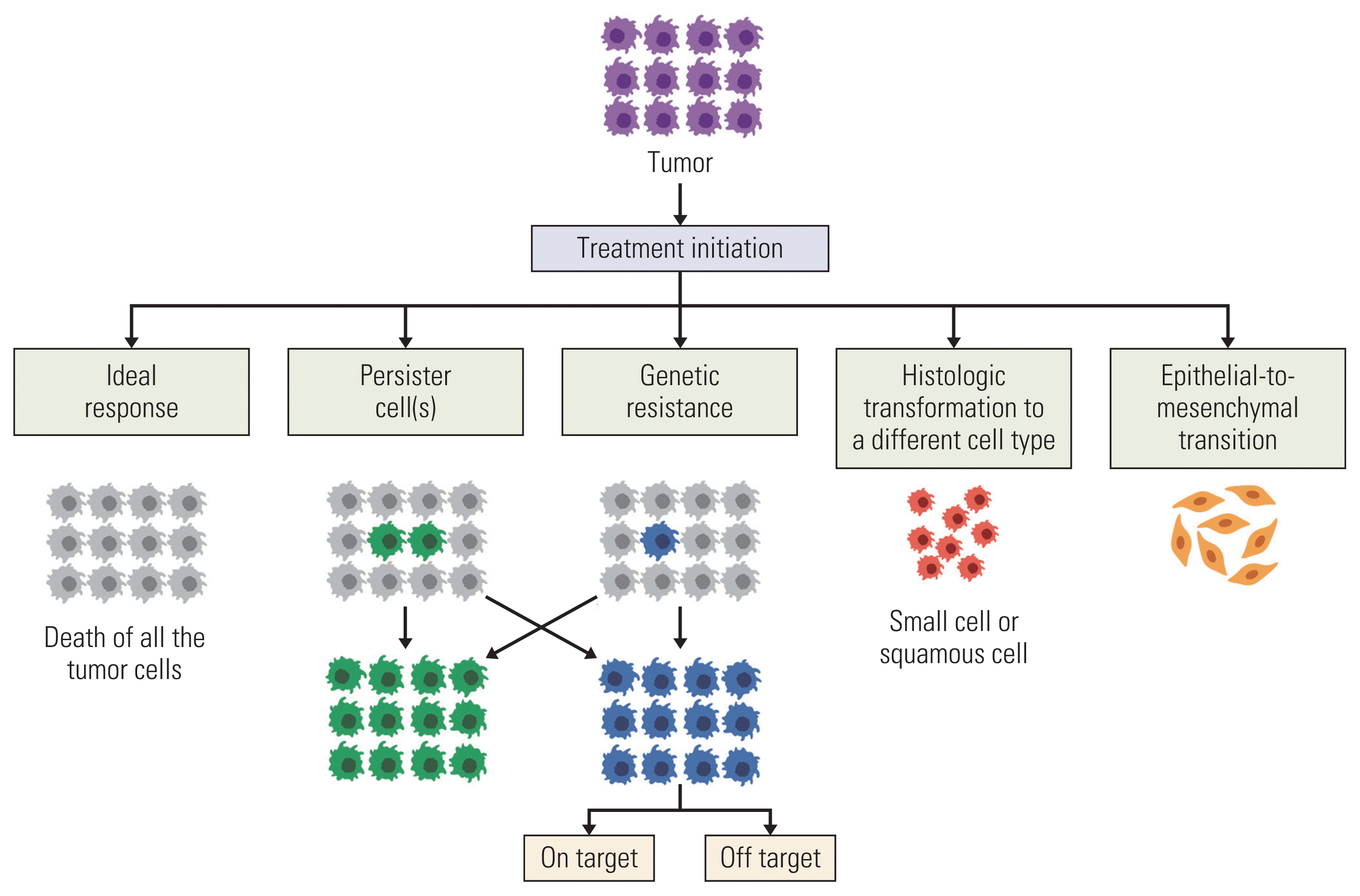
Many mechanisms of acquired resistance have been recognized (Table 2, Fig. 1) [47–57]. They may be divided into: (1) driver oncogene alterations, (2) abnormal signaling pathway alterations in parallel or downstream fashion, (3) pro-survival signaling through a different or unknown signaling pathway, (4) histological transformation from one cell lineage to another, and (5) drug tolerant cancer persister cells [58,59].
(1) Driver oncogene alterations
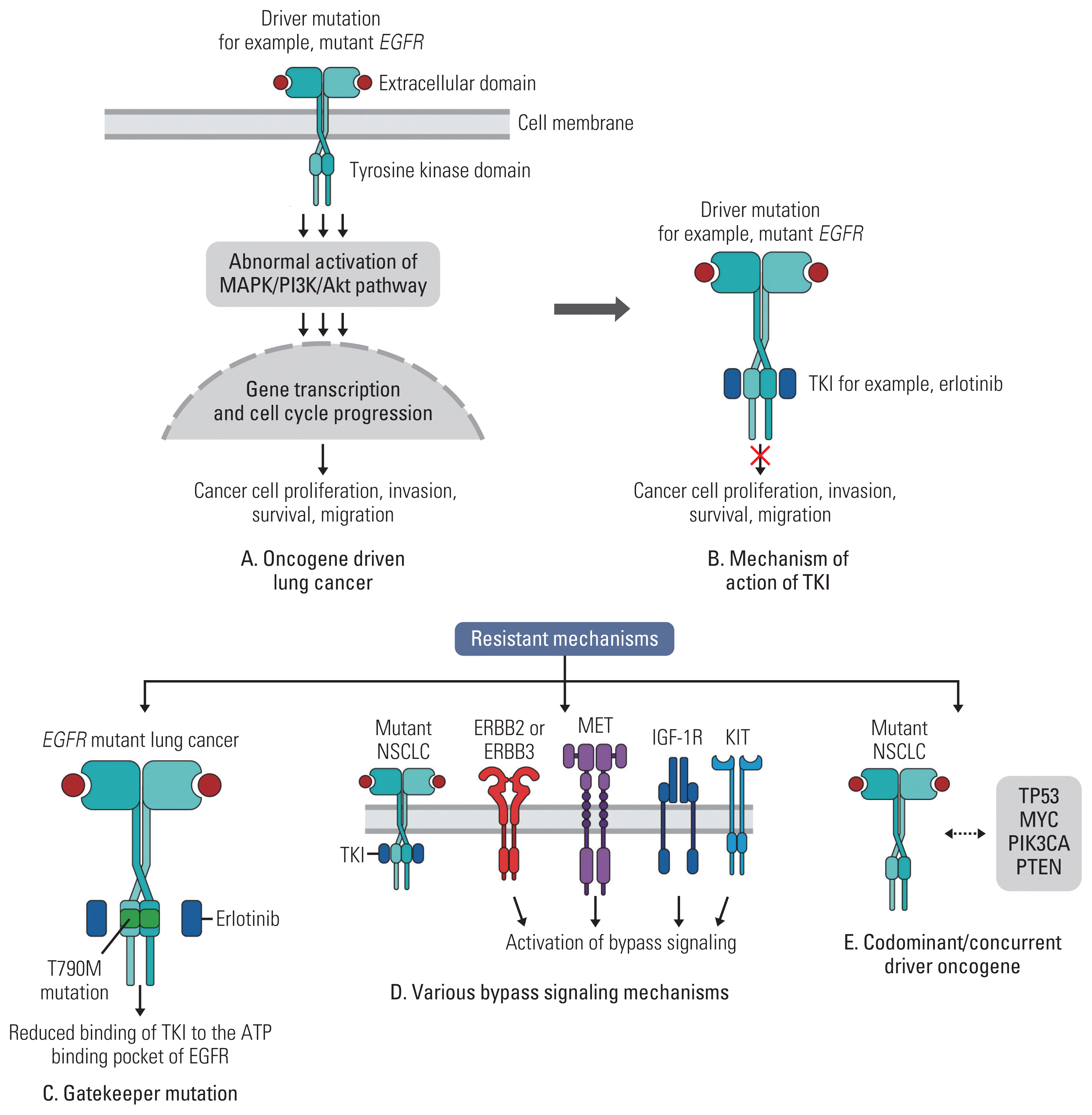
In certain situations, there are alterations in the target gene, such as mutations and amplifications that allow cells to proliferate even in the presence of an inhibitor. Targeted therapies can impose selective pressures causing aberrancies that activate the reengagement of the driver oncogene. Malignant tumors bearing specific aberrancies have significant initial responses to selective inhibitors. Unfortunately, resistance tends to develop over time. Examples of genetic aberrations in the target gene as a mechanism of resistance include secondary mutations after therapy with osimertinib such as EGFR C797S [60]. As well, on initial therapy with erlotinib or gefitinib, EGFR T790M mutations have been found to confer resistance [47].
(2) Off-target resistance
Off-target resistance relates to abnormal activation of: (1) downstream or parallel bypass signaling, (2) presence of co-dominant driver oncogenes, (3) lineage plasticity, (4) epithelial-mesenchymal transition (EMT), and (5) persister cancer cells [61] (Figs. 1 and 2).
1) Downstream or parallel bypass signaling
There are three major oncogenic signaling pathways that drive cell growth, proliferation and metabolism of cells: the PI3K/AKT/mTOR (PI3K pathway), RAS/RAF/ERK (MAPK pathway), and STAT/JAK pathways [7] (Fig. 2).
Some of the most common abnormal signal activation processes follow gain-of-function mutations, genomic amplification, chromosomal rearrangements, or autocrine activation. A gain-of-function mutation develops when downstream signaling is abrogated despite upstream blockade by targeted agents. Gene amplification is an increase in the number of copies of a gene sequence. It occurs in cancer cells when multiple copies of genes are produced in response to signals from other cells or their environment [62]. Chromosomal rearrangements can lead to cancer either by forming a hybrid gene or by causing dysregulation of a gene [63]. Autocrine signaling is defined as the production and secretion of an extracellular mediator by a cell followed by the binding of that mediator to receptors on the same cell to initiate signaling [64].
In BRAF mutant malignancies, treatment with BRAF inhibitors lead to amplification or activating of mutations that restore downstream MAPK signaling [65]. In NSCLC, adding a downstream MEK inhibitor to BRAF inhibitors improved duration of responses from 6.3 months to 10.2 months [26,66].
MET amplification increases sensitivity of hepatocyte growth factor signals and increases flux through the PI3K/AKT-mTOR axis, thus bypassing EGFR TKI inhibition [65]. This is the most common “bypass” resistance mechanism to osimertinib [60]. On the other hand, amplification of ERBB2 is more common resistance mechanism to the first-generation and second-generation EGFR TKIs [67]. Subsequent studies identified other mediators of parallel survival pathway activation in EGFR-mutant lung cancer, including FGFR3 amplification, PIKC3A mutations, and AXL overexpression all of which reactivate downstream MAPK and PI3K signaling [65,68–70].
A resistant mechanism worth mentioning is AXL overexpression. AXL is a receptor tyrosine kinase implicated in cellular proliferation and differentiation, cytoskeletal rearrangement, migration, and survival signaling [71]. AXL is known to mediate biological variables that function as modifiers of therapy resistance such as EMT, the tumor microenvironment and tumor heterogeneity. Intratumor AXL is involved in both intrinsic and adaptive resistance mechanisms in NSCLC. A retrospective study by Taniguchi et al. [72] demonstrated that higher AXL expression negatively correlated with EGFR TKI response, including osimertinib. AXL receptor upregulation results in acquired resistance even in the absence of the EGFR T790M mutation. Inhibition of AXL activation has shown effectiveness in overcoming acquired resistance to EGFR TKI treatment in NSCLC [72]. Dubermatinib (TP-0903) is a small molecule AXL inhibitor. A phase I study of 36 patients demonstrated manageable safety and early signs of biologic activity [73]. Antibody drug conjugates targeting AXL are also in clinical trials. The relation between AXL and other oncogenes (e.g., MET, BRAF, EGFR, ALK) is complex and heterogeneous. Understanding interactions between AXL and other oncogenes could help shape the development of new drugs.
2) Co-dominant driver oncogenes
While apparently single oncogenic drivers have been identified and targeted in NSCLC, it has become clear that a spectrum of non-sensitizing mutations co-exists and these lead to disease resistance to targeted therapies. Various studies have demonstrated inferior outcomes (decreased responsiveness, shorter progression-free survival [PFS] and overall survival [OS]) in patients with TP53 co-mutations in EGFR-mutant NSCLC with TKI therapy [74–76]. This is also seen with other mutation subtypes, including ALK and ROS-1 alterations [77–79]. Recently, Yu et al. [74] molecularly profiled EGFR-mutant tumors before and after disease progression on TKI therapy and demonstrated that in addition to TP53 mutations, concurrent HER2 amplification and MET amplifications were associated with shorter time to disease progression. Additionally, Alidousty et al. [80] demonstrated that the co-occurrence of TP53 mutations in ALK rearranged NSCLC can potentially lead to chromosomal instability leading to amplifications of other known cancer genes in one-fourth of TP53 mutated cases. For instance, 14% of TP53 mutant cases had MYC amplifications. MYC-overexpressing tumor cells have a high proliferative advantage compared to wild-type and this could potentially lead to drug resistance and disease progression.
3) Tumor lineage plasticity
Lineage plasticity is the ability of a cell to transform to a new phenotype that more closely resembles a distinct developmental lineage [81]. Cancer cell plasticity occurs after exposure and selective pressure to targeted therapy agents [81,82]. This transformation allows the tumor cells to adapt to adverse conditions, including a hypoxic tumor microenvironment [81]. Although these transformed cells retain the driver mutation, they are no longer dependent on the driver mutation for proliferation causing therapeutic resistance.
In NSCLC, histologic transformation from adenocarcinoma to small-cell or squamous cell lineage has been identified [83]. These changes have been described in lung adenocarcinoma with EGFR mutations, EML4-ALK fusions and KRAS G12C mutations treated with targeted therapies [84–88].
This switching phenomenon may occur in up to 15% of cases of EGFR-mutated lung adenocarcinoma with acquired resistance [60]. The presence of RB1 and TP53 mutations at baseline are a predictor of small cell lung cancer (SCLC) transformation [81,89]. DNA sequencing of neuroendocrine transformed cells indicates that tumor cells retain the EGFR mutation, suggesting a clonal evolution from adenocarcinoma to SCLC [90,91]. To date, there are no predominant genomic correlations associated with EGFR-mutated adenocarcinoma to squamous histology transformation. Addressing the epigenetic, genomic and microenvironmental factors that drive lineage plasticity will be pivotal for the development of new treatment strategies.
4) Epithelial-mesenchymal transition
EMT is a biological process in which epithelial cells lose their characteristics and acquire mesenchymal features [61]. This process has been implicated in lung tumor progression and metastasis [92]. It is believed to occur by stimulation and activation of intracellular signaling pathways that lead to the downregulation of E-cadherin.
Evidence suggests that EGFR TKIs can cause cells to convert to a mesenchymal phenotype by the decreased expression of E-cadherin, the expression of N-cadherin and other mesenchymal markers [93]. NSCLC cells with acquired resistance to gefitinib or osimertinib have shown EMT characteristics, with a decrease in E-cadherin, and increases in vimentin and stemness, without any secondary EGFR mutations [93,94]. Interestingly, AXL is implicated in the pathogenesis of EMT and preclinical data indicate that AXL inhibitors can restore sensitivity of mesenchymal EGFR-mutant cells to erlotinib [95].
5) Persister cancer cells
The concept of drug tolerant persister cells (DTP) has gained recent attention. Persister cancer cells are defined as a population of cells that survive systemic treatments by entering a reversible and slow proliferation state [96,97]. These cells are thought to be different from cancer stem cells (CSCs). Persister cells tend to lack classic drug resistance driver alterations and their resistant phenotype could be transient and reversible upon removal of the treatment drug [98]. In contrast, CSCs are cells within a tumor with the ability to self-renew and generate heterogenous lineages of cancer cells [99]. Despite standard of care treatments, persister cells survive and eventually develop resistance pathways. This leads to abnormal survival and cell growth in relapse sites.
The survival of persister cells is thought to occur due to pre-existent drug-resistant cells or by induction of intrinsic changes that promote phenotypic variations. As malignant cells proliferate, they are subjected to different pressures, metabolic, hypoxic and nutritional limitations [59]. A series of case reports show how EGFR inhibitors induce chromatin repression, adding to mechanisms that influence the development and survival of DTPs [97].
New studies are focused on identifying, understanding, and targeting DTPs. There is increasing interest in eliminating, reducing, or suppressing DTPs as a critical step for improving response and clinical outcomes [59,98]. Other strategies in oligometastatic disease may follow local control by surgical resection or radiation therapy, as they have shown to prolong PFS in small studies [100].
Sequential tissue biopsies combined with genetic sequencing may allow for better characterization of evolving tumor characteristics. There is a clear need of understanding the role of DTP in NSCLC and its role in the design of treatment strategies
Toxicity
Targeted therapies are designed to disrupt genetic aberrancies that act as drivers in cancer development. Although quite tolerable after short term exposures, because they tend to be used for chronic continuous administration, they come at the expense of significant toxicities. Targeted agents were initially presumed to be relatively non-toxic since they abrogate cancer specific targets. However, their “on target” and “off target” effects can lead to toxicity. Either of these adverse reactions may prevent the recommended schedule and dose of therapy, thus potentially limiting drug efficacy. Common adverse events of target therapies are summarized in Table 1 [6,13–35].
1. On-target toxicity
On-target toxicity occurs when a protein is inhibited in normal cells. Targeted agents were initially presumed to be relatively non-toxic since they were designed to abrogate cancer specific targets. However, because of complexities of cell signaling and our agents not being able to strictly and selectively block aberrant proteins in the cancer cells, inhibiting a fixed set of proteins in the normal cells causes toxicity by inhibiting the signaling pathway. These on-target toxicities exhibit a class effect where all the drugs in the same class have similar toxicity. For instance, hyperglycemia with PI3K-inhibition, hypertension with vascular endothelial growth factor inhibition and skin rash with EGFR inhibition [101–103].
As mentioned above, EGFR inhibitors, as a class, are associated with cutaneous side effects such as rash, dry skin and pruritus. These events are thought to occur due to the high expression of EGFR in the epidermis and the release of inflammatory cytokines by EGFR inhibitors [103,104]. These symptoms generally appear within the first week of treatment and peaks in intensity around the second week of therapy. Third generation inhibitors are more selective for mutant EGFR. Thus, skin rash associated with EGFR inhibition by osimertinib is much milder as compared with first-generation EGFR TKIs.
(1) Off-target toxicity
Off-target toxicity occurs when a particular drug blocks a non-target protein leading to a deleterious side effect. This, as a rule, does not exhibit a class effect and is thought to be related to the pharmacology of the specific drug. One of the classical examples is the cardiac toxicity which is associated with osimertinib but not with gefitinib. Cardiovascular toxicity associated with osimertinib include heart failure, left ventricular dysfunction, conduction abnormalities, and myocardial injury or dysfunction [105,106]. EGFR is a receptor tyrosine kinase in the ErbB/HER family which also includes HER2. Osimertinib has modest activity against HER2, possibly explaining its cardiotoxicity [107].
Factors Influencing Toxicity
The broad types of toxicity from targeted agents have been described above. Apart from the inherent drug characteristics leading to “on-target” and “off-target” toxic effects, a number of factors contributing to toxicity have been recognized. These are described below.
1. Age
On average, NSCLC is diagnosed in patients older than 65 years [108]. Treating this sub-group of patients with multiple co-morbidities, who tend to be on multiple drugs is a challenge. More importantly, they tend to be underrepresented in pivotal trials.
In a phase III study, a geriatric population with advanced EGFR-mutant NSCLC and treated with erlotinib versus placebo was evaluated for treatment toxicities. Elderly (> 70 years of age) patients treated with erlotinib experienced greater toxicity: 35% versus 18% (grade > 3; p < 0.001) [109]. The increased toxicities reported in the elderly included rash, stomatitis, dehydration, anorexia, and fatigue. Response rates were similar between age groups, but at the cost of increased toxicity.
A retrospective multicenter study evaluated patients > 80 years old with EGFR mutations to assess efficacy and tolerance of TKIs [110]. The overall response rate and disease control rates were 63.3% and 78.9%, respectively. Median PFS and median OS were 11.9 months and 20.9 months, respectively. The predominant toxicities were cutaneous for 66.7% of patients (grade 3–4, 10%), diarrhea for 56.0% (grade 3–4, 15%; grade 5, 2%) and others for 25.7% (grade 3–4, 41%). Octogenarians with EGFR-mutated NSCLC treated by EGFR TKI had clinical outcomes and toxicity profile comparable to younger patients.
In summary, it would appear that older patients with oncogene driven tumors derive the same benefit as younger patients but with increased toxicity. There have been few studies in this population, but toxicities may be related to diminished organ function. With the aging of the population, it is imperative that studies aimed at understanding mechanisms of toxicity in older adults are performed so that rational approaches to dose adjustments can be defined.
2. Central nervous system toxicity of brain penetrant targeted therapies
In the development of targeted agents for NSCLC, subsequent generations of drugs in each class tend to be more selective with increased central nervous system (CNS) penetration. One of the major concerns with the CNS penetrant drugs has been concomitant CNS toxicity. For instance, lorlatinib, entrectinib and larotrectinib are TKIs with good CNS activity but do have significant CNS toxicity [22,28,111–113]. This creates a challenge in the management of these patients and frequently requires dose interruption and dose reduction. This is further complicated by the fact that a significant number of NSCLC patients with driver mutations will develop CNS metastases [114–116]. Further research is needed to develop CNS penetrant drugs with decreased CNS toxicity.
3. Intermittent scheduling
There are a few cancer types where metastatic disease is curable with systemic therapy. In almost all these cases such as lymphoma, testicular cancer, germ cell tumors, leukemia, Wilms tumors and retinoblastomas, combination therapy is used. The objective of systemic cancer therapy is to reduce the tumor cell population to zero.
Curative systemic therapy generally comprises a combination of agents delivered cyclically at the highest tolerable dose. The Goldie-Coldman hypothesis suggests that tumors undergo a spontaneous mutation of about one cell in 105 cells per gene [117]. At the time of detection, with current imaging, most tumors are about 1 gram, containing 109 cells and, consequently, about 104 resistant clones to a given drug. Resistance to two drugs, however, should be seen in one cell out of 1010 cells. It, therefore, follows that multidrug therapy will be more effective than single-agent therapy. In addition to overcoming tumor resistance, combination therapy is important in limiting drug toxicity, since several agents with non-overlapping toxicities can be utilized [118].
In spite of the above well-known principles, attempts to combine targeted therapies inhibiting complimentary pathways to prevent resistance and potentially achieve cures have been unsuccessful, because of toxicity. Examples include combinations of EGFR TKIs and mTOR inhibitors, EGFR TKIs and insulin-like growth factor 1 receptor inhibitors, MEK inhibitors and PI3K inhibitors among others [119].
In almost all of these studies, the agents have been dosed continuously. An approach to minimizing toxicity could be evaluation of intermittent dosing as well as sequential schedules, based on robust preclinical data.
4. Drug-drug interactions
Drug interactions that can lead to reduced or increased plasma levels of targeted agents can lead to reduced efficacy or increased toxicity.
Smoking has been reported to alter the metabolism of some targeted agents. In a reported study, current smokers had significantly less erlotinib exposure following a single 150 or 300 mg dose than nonsmokers. Results showed increased metabolic clearance of erlotinib in current smokers [120].
Studies have demonstrated pH-dependent solubility with gefitinib and erlotinib and hence co-administration with H2-blockers or proton pump inhibitors is known to reduce drug exposure and efficacy [121,122].
Additionally, with TKIs which are predominantly metabolized by CYP3A (erlotinib, gefitinib, afatinib, dacomitinib, crizotinib, ceritinib, lorlatinib and dabrafenib), concomitant use of strong CYP3A inhibitors or inducers may increase or decrease the efficacy/toxicity of the TKI respectively [122–124]. Of note, grapefruit is a strong CYP3A inhibitor and St. John’s wort is a strong CYP3A inducer.
5. Food effect
Most available targeted agents are administered orally. Oral bioavailability is determined by absorption and first-pass metabolism [125]. Food can affect drug bioavailability when fat content from food increases bile salt secretion. This in turn increases solubilization and absorption of lipophilic drugs. Also, food substances can bind to the oral drug and limit its absorption [125]. Other factors that can either increase or decrease absorption include a rise in gastrointestinal pH, delay in gastric-emptying rate and increase of splanchnic blood [125–127].
In many clinical trials, drug administration is provided on an empty stomach. This is usually recommended to reduce variability of drug absorption between patients. An example of food effect is seen with osimertinib. The Cmax and area under the curve are increased by 14% and 19%, respectively, following a high-fat, high calorie meal when compared to fasting [128]. Food also increases the steady state levels of erlotinib [129]. How different foods and acid reducing agents affects the absorption and efficacy of each targeted therapy needs to be further explored.
Pharmacogenomics
Pharmacogenomics is an evolving branch of pharmacology and seeks to understand the association between genetics, response rates and toxicities [130]. It is an individual variation in drug toxicity and efficacy related to inherited variations in drug transport, metabolism and target enzymes. In a number of cases, specific genetic polymorphisms may be more prevalent in specific ethnic populations. For example, ethnic differences in the distribution of EGFR polymorphisms have been described [131]. Additionally, ethnicity may play a role in drug tolerability. For instance, rate of interstitial lung disease with gefitinib or erlotinib is significantly higher in Japanese population [132]. This example exposes the need of expanding ethnic diversity in clinical trials and understanding the role of germ-line polymorphisms in drug metabolism enzymes in drug toxicity [133,134].
A major challenge to incorporating pharmacogenetics into routine NSCLC care include establishing validation and standardization of genotyping procedures to consistently include the genotyping of normal tissue from each patient and not focus only on tumor tissue [130]. This could help identify germ-line polymorphisms of clinical relevance in each patient.
Integrating pharmacogenetics in NSCLC is an important step in achieving the best personalized medical care, leading to optimizing drug efficacy and minimizing toxicity.
Cost and Access
Cancer drugs are very expensive in the United States (see Table 1 for drug prices). Even though there are multiple drugs in each class targeting an oncogene product, in-class competition does not appear to put a downward pressure on prices as happens in other areas of the economy, and prices continue to remain high [135,136]. Since global pricing follows the United States pricing, targeted drugs for oncogene-addicted NSCLC remain high globally and, in most countries, these agents are unaffordable to the majority of the NSCLC population [136,137].
Conclusion
A new generation of drugs is replacing the chemotherapy-based standard of care in systemic therapy of NSCLC. As with all important innovations, targeted therapies pose unique challenges. An approach to overcoming both primary and acquired resistance in oncology is combination therapy, where drugs inhibiting different pathways are combined to achieve maximum response and prevent the emergence of resistance. This approach has led to the cure of some advanced cancers such as lymphoma and testicular cancers. Because of the chronic oral administration, toxicity of targeted therapy continues to be a major issue which precludes the use of these agents in combination and thus limits the opportunity for cure. Future studies should focus on understanding the mechanisms of toxicity, developing novel drug administration schedules to allow for combination of TKIs targeting complimentary pathways. Studies should also focus on special populations and host factors contributing to variations in toxicity. The lack of affordability of targeted agents by most lung cancer patients in the world is a concern. Global efforts to reduce drug prices is an urgent need.
 XML Download
XML Download