

 PDF
PDF Citation
Citation Print
Print



Introduction
Members of the RAS family are involved in the regulation of many signal transduction processes in normal cells. When RAS mutated, they are persistently activated and promote oncologic events, including the transcription of cancer-related genes, and cell proliferation and survival. During the last three decades however, ongoing efforts to treat RAS-mutant tumors have failed due to the inability to target the RAS oncoprotein itself. This is due to its high affinity for GTP, and its lack of a clear binding pocket due to blocking by the enzyme’s farnesyltransferase and geranylgeranyl transferase. These enzymes are required RAS prenylation or the lipid posttranslational modification needed for the proper cellular localization of RAS family members, particularly KRAS and NRAS [1]. Alternatively, as mutated KRAS persistently activates its downstream signaling pathways, such as RAF/MEK (mitogen-activated protein kinase/ERK [extracellular-signal-regulated kinase])/ERK or phosphatidylinositol 3-kinase/AKT (protein kinase B)/mammalian target of the rapamycin, agents blocking one or more downstream signaling factors could potentially be used to overcome the targeting failures. Notably in this regard, we reported previously that such a blockade was efficacious at first but subsequently became ineffective soon through activation or bypass mechanisms involving alternative pathways [2,3]. Moreover, our attempts to enhance the efficacy of this approach by blocking more targets in downstream signaling pathways induced severe or unacceptable toxicity levels as members of the RAS family have many important functional roles in normal cells.
Fortunately, however, some recent creative approaches have revealed new binding pockets on the surface of the KRAS protein, thereby presenting potential new targeting options. Among these new possibilities, the covalent targeting of mutant KRAS, blockade of KRAS interactions with associated proteins indispensable for membrane associations, and inhibition of KRAS-driven malignant phenotypes and KRAS synthetic lethal interactions, have shown some promise as effective therapeutic pathways for KRAS-mutant tumors [4,5]. Sotorasib (AMG-510), the first new agent of this type that targets the KRAS G12C mutation, has shown clinical efficacy in solid tumors. In the non–small cell lung cancer (NSCLC) subgroup of cancers, the response rate and median progression-free survival was 32.2% and 6.3 months, respectively, to this drug while the outcomes with colorectal cancer patients were 7.1% and 4.0 months, respectively [6]. Adagrasib (MRTX849), another agent that targets KRAS G12C, has also initially shown promising clinical activity with a response rate of 45% against NSCLC, and of 17% against colorectal cancer [7,8]. The target of these agents is limited to tumors harboring the KRAS G12C mutation however and their clinical efficacy still seems modest when compared with EGFR or ALK tyrosine kinase inhibitors. This indicates that we need new therapeutic approaches covering different types of KRAS mutations to achieve a higher clinical efficacy of targeting agents.
Heat shock protein-90 (HSP90) has remained a viable anti-cancer target as it acts as a molecular chaperone that stabilize numerous oncogenic proteins, such as EGFR, ALK, BRAF, HER2, AKT, and MEK, that play major roles in tumor proliferation and survival. In addition, a relationship between HSP90 expression and the prognosis in cancer patients has been reported [9]. Geldanamycin [10] and radicicol [11], first-generation HSP90 inhibitors exert their anti-cancer effectiveness by pharmacologically destabilizing multiple KRAS signaling molecules [12] and subsequently, second- and third-generation HSP90 inhibitors have developed. However, despite their promise in preclinical studies, HSP90 inhibitors have displayed limited or disappointing clinical activity. In many clinical studies, these drugs achieved stable disease at best or short-lasting efficacy due to the onset of resistance. A variety of chemically potent and selective HSP90 inhibitors has been developed to date and advanced to various stages of a clinical trial [13]. Among these agents, a third-generation HSP90 inhibitor targeting KRAS-mutated NSCLC tumors, AUY922, was developed via a collaboration between the Institute of Cancer Research and Vernalis and was licensed to Novartis. It was withdrawn in 2012 however by Novartis after failing to show efficacy in a clinical trial as a single agent [14]. HSP90 inhibitors are still at various stages of preclinical and clinical development however as counterparts for combination therapies [9,15]. A combinational approach might be required to overcome the limitation of HSP90 inhibitors and resistance mechanisms would need to be well characterized to find the best partners. An understanding of the underlying mechanism will also be very helpful for developing new HSP90 inhibitors [13].
MicroRNAs (miRNAs) are well known as small non-coding RNAs that regulate gene expression. A lot of studies have shown that miRNA expression is overexpressed or deregulated in most cancer [16]. In addition, there are numerous research articles regarding the functions of miRNAs in the development of drug resistance [17]. Therefore, miRNA-based anti-cancer therapies are being developed either alone or in combination with current targeted therapies as anti-cancer sensitizer.
In our present study, we investigated the mechanism of resistance to AUY922, to explore possible avenues of overcoming and expanding the field of HSP90 inhibitors as targeted cancer drugs and we tried to identify novel resistance biomarkers, including proteins and miRNAs, of HSP90 inhibitor and also provide some insights that may assist with the future development of successful next-generation HSP90 inhibitors.
Materials and Methods
1. Cell culture and reagents
The human KRAS-mutated NSCLC cell lines, H23, H358, H647, H1944, and A549, were purchased from the American Type Culture Collection (Manassas, VA) and grown at 37°C in 5% CO2 using RPMI-1640 (Welgene Inc., Gyeonsan, Korea) containing 10% fetal bovine serum (FBS) and 1x penicillin-streptomycin solutions from Welgene Inc. AUY922 (HSP90 inhibitor, luminespib), cilengitide trifluoroacetate (inhibitor of the αvβ3 receptor), and TAE226 (focal adhesion kinase [FAK] inhibitor) were purchased from Selleck Chemicals (Houston, TX). These agents were each dissolved in dimethyl sulfoxide to a final concentration of 10 mM and stored at −20°C.
2. Cell viability assay
Cell viability was measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI) in accordance with the instruction manual. Briefly, 3×103 cells were plated in triplicate wells in 96 microtiter plates in a volume of 90 μL. On the following day, the cells were incubated with the desired concentrations of AUY922 and/or TAE226 and/or cilengitide to a final volume of 100 μL. After 72 hours, 100 μL of CellTiter-Glo reagent was added and the cells were then incubated for 10 minutes at room temperature. The luminescence levels were measured using a Wallac 1420 (PerkinElmer, Boston, MA).
3. Establishment of AUY922-resistant cells
To establish resistant cells, AUY922 treated to A549 and H1944 cells by continuous exposure to increasing concentrations of AUY922 (2, 5, 10, 20, 40, 80, 160, 320, 500, 750, 1,000 nM). Up to 160 nM, incubate cell for 4 days in each dose and up to 1,000 nM, incubate cell for 6–7 days in each dose.
4. Gene expression profiling by RNA-seq
To discover differentially expressed genes (DEGs), we compared mRNA expression profiles between both two AUY922 resisstant cell lines. RNA samples were purified using the RNeasy kit (Qiagen, Hilden, Germany). Biotinylated cRNA was prepared using the Illumina RNA Amplification Kit (Ambion, Austin, TX) according to the manufacturer’s directions starting with approximately 500 ng total RNA. Hybridization to the Sentrix HumanRef-8 Expression BeadChip (Illumina), washing and scanning were performed according to the Illumina BeadStation 5006 manual (revision C). Array data processing and analysis was performed using Affymetrix Expression console1.4, R program (3.1.3), DAVID 6.7. Expression changes for individual genes were considered significant if they met four criteria: z-ratio above 1.4 (or below −1.4 for downregulated genes); false detection rate < 0.30; p-value of the pairwise t-test < 0.05; and mean background-corrected signal intensity z-score in each comparison group is not negative. This approach provides a good balance between sensitivity and specificity in the identification of DEGs, avoiding excessive representation of false positive and false negative regulation.
5. Establishment of an integrin αvβ3 stable cell line and miR-150/miR-142 overexpressing stable cell line
The full-length cDNA for the integrin β3 (ITGB3) subunit was purchased from Thermo Fisher Scientific (Waltham, MA) and cloned into the pLVX-IRES-Puro vector (Clontech, Mountain, CA) using XbaI and XhoI restriction sites. For ITGB3 overexpression, the cDNA of ITGB3 was cloned into the pcDNA3.0 vector. The integrin αv (ITGAv) gene was first polymerase chain reaction (PCR) amplified from cDNA and then subcloned into the hemagglutinin pcDNA3.0 plasmid to obtain the ITGAv expression vector. This PCR was performed using the following primers: sense, 5′-CGG GAT CCA ATG GCT GCT CCC GGG-3′, and antisense, 5′-ATT TGC GGC CGC TTA GGT TTC AGA GTT TCC TTC G-3′, and the following cycling conditions: 94°C for 3 minutes followed by 35 cycles of 94°C for 30 seconds and 48°C for 3 minutes, followed by 72°C for 10 minutes. RNA was extracted using a RNeasy Mini Kit (#74104, Qiagen) in accordance with the manufacturer’s instructions. The RNA was then reverse transcribed via a High-Capacity cDNA Reverse Transcription Kit (#4368814, Thermo Fisher Scientific). cDNAs were obtained using a QIAquick Gel Extraction Kit (#28704, Qiagen). To establish stable cell lines, the pcDNA3.0/ITGAv and pcDNA3.0/ITGB3 or -miR-150-5p (HmiR0306-MR04, GeneCopoeia, Rockville, MD) and -miR-142-5p (HmiR0282-MR04, GeneCopoeia) expression vectors were transfected using lipofectamine 2000 plus (Invitrogen, Carlsbad, CA), following the manufacturer’s instructions. All vectors were linearized using the AhdI (New England Biolabs, Ipswich, MA) enzyme to increase the efficiency of plasmid integration. Cells (at 70%–80% confluence) were washed with Opti-MEM (Invitrogen), and incubated with a DNA-lipofectamine mixture (2 μg DNA and 3.5 μL lipofectamine reagent). For stable transfection, the transfected cells were selected with 500 μg/mL of the antibiotic G418 (Welgene) or 1 μg/mL of the puromycin (Welgene). Parental cells transfected with empty vectors were used as controls. Integrin αvβ3 (ITGAvβ3) overexpression was detected by western blotting using an anti-human integrin β monoclonal antibody (#13166, Cell Signaling Technology, Danvers, MA) and ITGAv (D2N5H) monoclonal antibody (#60896, Cell Signaling Technology). miR-150 (A25576, hsa-miR-150-5p, 477918_mir, Thermo Fisher Scientific) and miR-142 (002248, hsa-miR-142-5p, 4427975_mir, Thermo Fisher Scientific) expression were measured by reverse transcription polymerase chain reaction (RT-PCR).
6. Quantitative RT-PCR
Quantitative RT-PCR was performed using RNA isolated from NSCLC cells. Briefly, total RNA was extracted using an miRNeasy Mini Kit (217004, Qiagen), in accordance with the manufacturer’s instructions. cDNA was synthesized with a Taqman microRNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA). RT-PCR was performed on a LightCycler using SYBR Green (Roche, Basel, Switzerland) and a 7900HT Fast Real-Time PCR system (Applied Biosystems). Taqman gene expression assays (miR-150: hsa-miR-150-5p, A25576; miR-142: has-miR-142-5p, 4427975; ITGB3: Hs01001469_m1, 4453320; ITGAv: Hs00233808_m1, 4331182; GAPDH: Hs03929097_g1, 4453320) were purchased from Thermo Fisher Scientific. The comparative Ct method 2−ΔΔCt was used to calculate the changes in miRNA expression of all samples relative to a non-diseased sample, which was designated as the calibrator.
7. Western blot analysis
For western blotting, cell lysates were first prepared using RIPA buffer (Sigma-Aldrich, St. Louis, MO) in accordance with the manufacturer’s instructions. Protein samples were then applied to the wells of NuPAGE 4%–20% Tris-Gly gel, electrophoresed in sodium dodecyl sulfate running buffer (Invitrogen), and transferred to nitrocellulose membranes using the iBlot transfer apparatus (Invitrogen). Membranes were blocked in Tris-buffered saline containing 0.5% Tween 20 (TBS-T) and 5% bovine serum albumin (BSA) for 1 hour at room temperature, followed by incubation with the primary antibody overnight at 4°C. On the following day, after the membranes were washed three times for 10 minutes each in TBS-T, horseradish peroxidase–conjugated secondary antibody (Bio-Rad, Hercules, CA) in TBS-T containing 2% BSA was applied for 1 hour at room temperature. Proteins were visualized with ECL Plus enhanced chemiluminescence reagents (Amersham Biosciences, Piscataway, NJ) using G-box Chemi Systems (SynGene, Bengaluru, India). The commercial antibodies used in this study were ERK, phospho-ERK, MEK, phospho-MEK, AKT, phospho-AKT, c-Raf, tetraspanin 7 (TSPAN7), E-cadherin N-cadherin, cleaved poly(ADP-ribose) polymerase (PARP), PARP, ITGB3, ITGAv, HSP90, Phospho-p90RSK, RSK, phospho-FAK and β-actin (all purchased from Cell Signaling Technology).
8. Combination index analysis
Combination effects were evaluated using a CellTiter-Glo luminescent assay in cells treated with 1,000 nM AUY922 plus increasing concentrations of TAE226 (10, 100, 200, 500, and 1,000 nM) or cilengitide (10, 100, 200, 500, 1,000, 2,000, 5,000, and 10,000 nM). The fraction affected (Fa) and combination indexes (CI) were processed using CalcuSyn software (Biosoft). A CI of less than 1.0, equal to 1.0, or more than 1.0 were taken to indicate synergistic, additive, and antagonistic effects, respectively.
9. siRNA-mediated knockdowns
Human TSPAN7 proteins were knocked down in A549R cells using a final 15 nM concentration of two TSPAN7 siRNAs (Cat#4392420, s14203, s14204) sourced from Thermo Fisher Scientific. In parallel, a pool of Silencer Select Negative Control siRNA (also at a final 15 nM concentration) was used as negative control (4390843, ThermoFisher Scientific). For all siRNA transfection experiments, we used Lipofectamine RNAiMAX reagent (Invitrogen) in accordance with the manufacturer’s protocol.
10. Xenograft study
Six-week-old Balb/c-nu/nu female mice were purchased from Central Lab Animal Inc. (Seoul, Korea). An A549/ITGAvB3 xenograft model was established upon subcutaneous injection of 1×107 cells into the right flanks of these twelve animals. Ten days after inoculation, the mice were randomized into groups of four. NVP-AUY922 (10 mg/kg) was administered 3 days/wk and TAE226 (25 mg/kg) was administered 5 days/wk up to 21 days. Tumor sizes were assessed at least three times per week by caliper measurement and the lesion volumes were calculated using the following formula: tumor size (mm3)=(d2×D)/2, where d and D are the shortest and longest diameters of the tumor, respectively. Animal procedures were approved by the Asan Medical Centre Institutional Animal Care and Use Committee and Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines. p-values and percentage of tumor growth inhibition ratio (T/C) values were both calculated at the end of the experiment.
11. Migration and invasion assay
CytoSelect 24-Well Cell Invasion and Migration assay kits were purchased from Cell Biolabs (San Diego, CA). In accordance with the manufacturer’s instructions, cells were serum-starved overnight and 5×105 cells in 300 μL of medium without FBS were placed in the upper chamber of a transwell plate while the lower chamber was filled with 0.5 mL of medium supplemented with 10% FBS (Welgene). To quantify the number of migratory and invading cells, crystal violet-stained cells were obtained using an extraction buffer and subjected to spectrophotometric measurements at 560 nm.
Results
1. ITGAvβ3 induces AYU922 resistance via FAK activation
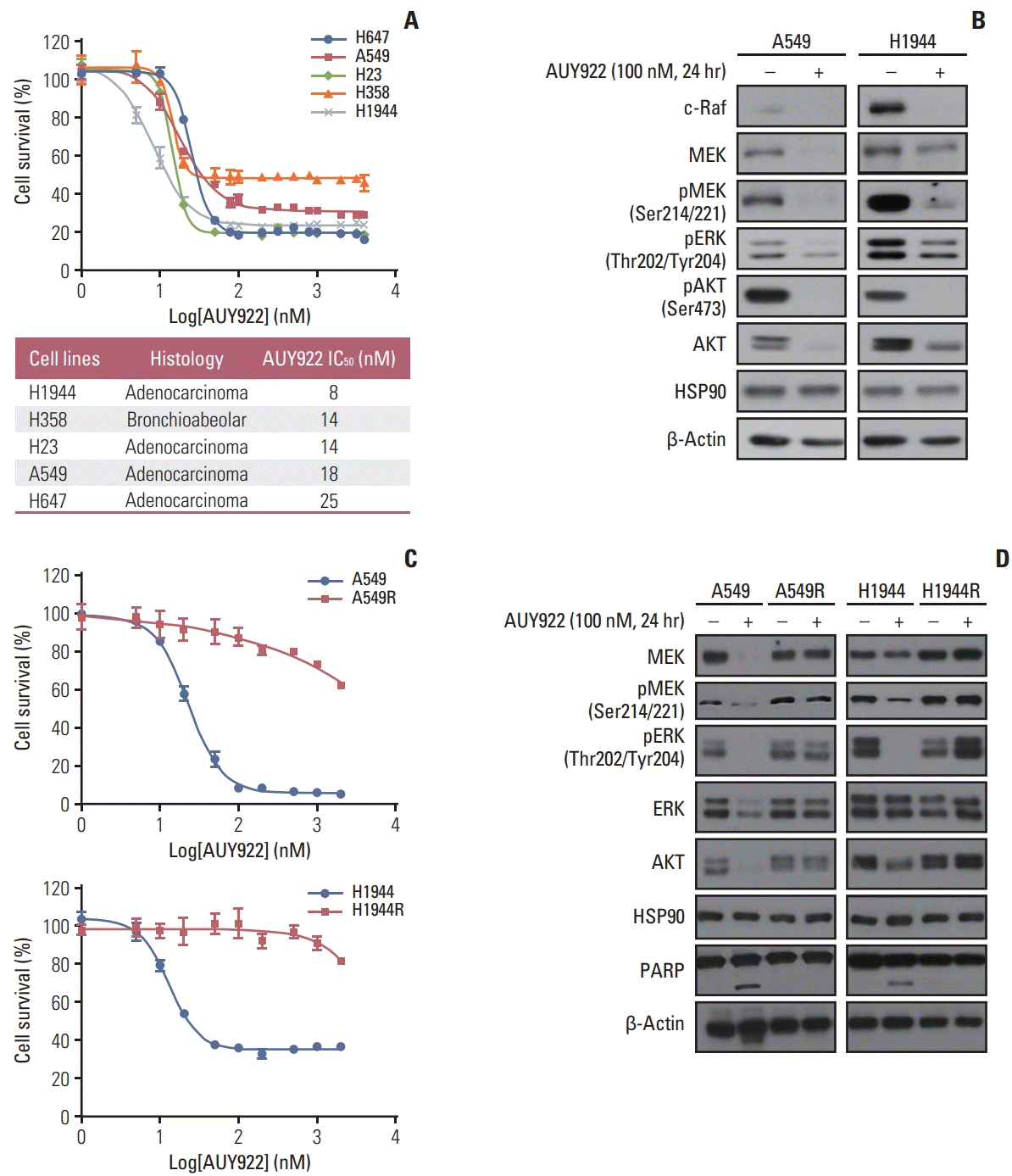
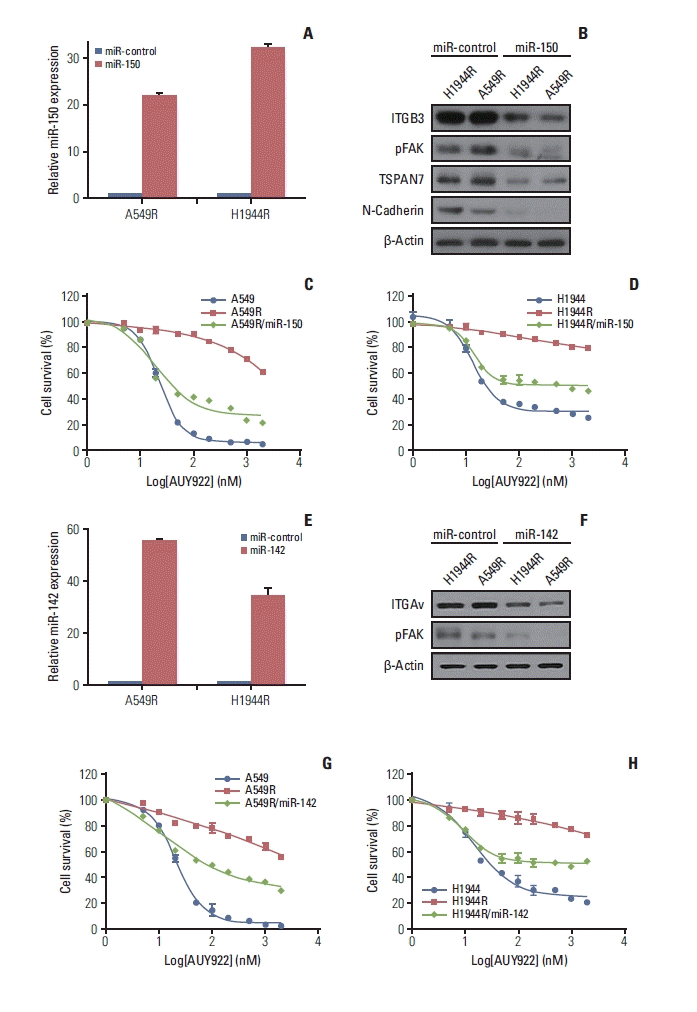
Most of the KRAS-mutant NSCLC cells tested in our current experiments were sensitive to AUY922 during the early treatment period (Fig. 1A and B). We selected two KRAS-mutated NSCLC cell lines, A549 and H1944, and treated them with increasing concentrations of AUY922 until they displayed resistance (Fig. 1C). AUY922 was eventually unable to fully block HSP90 activity in these two AUY922-resistant cell types, which we designated A549R and H1944R (Fig. 1D).
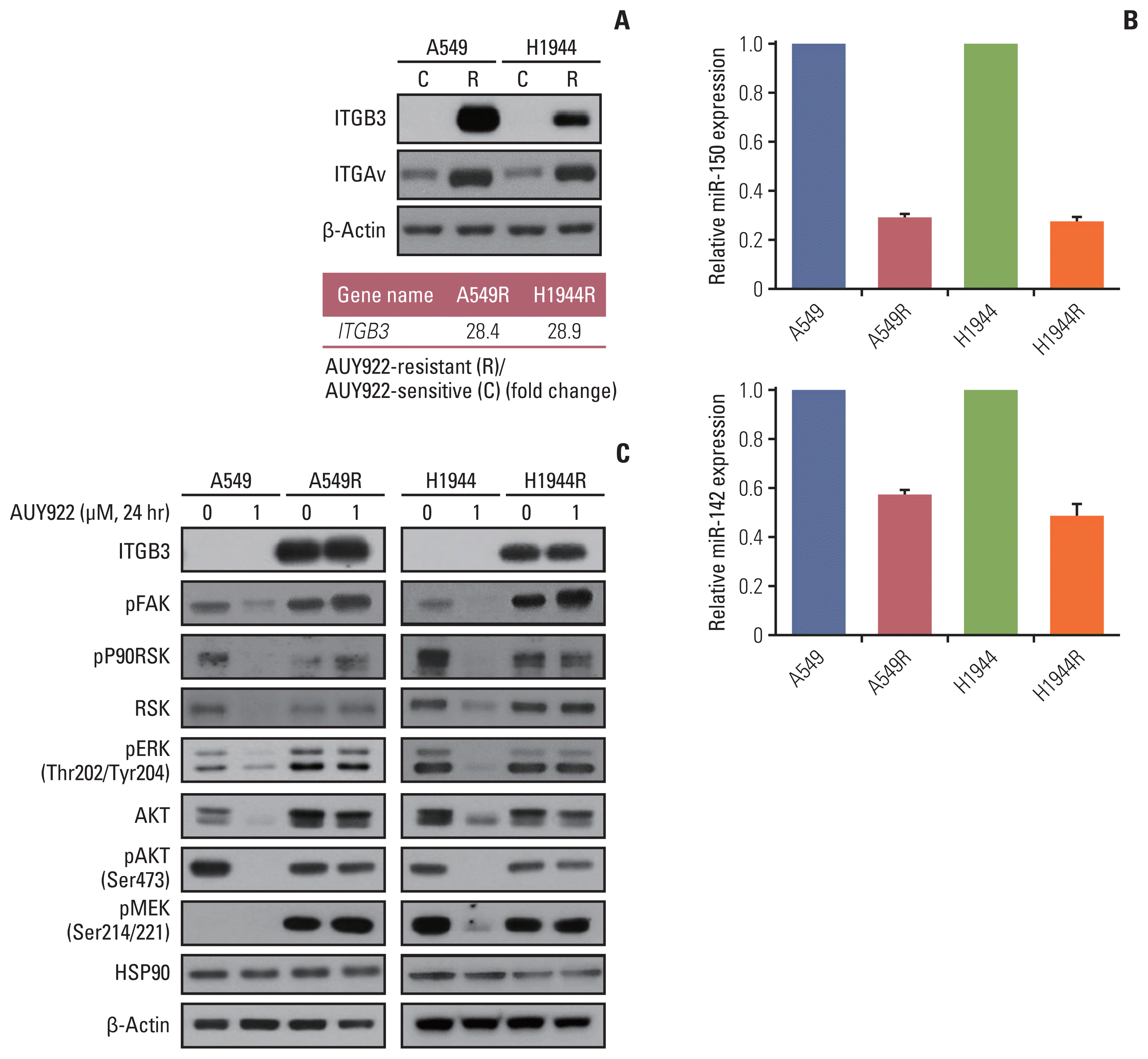
We conducted RNA sequencing analysis to identify DEGs between AUY922-resistant and parental sensitive cells. Among the candidate DEGs that were obtained, ITGB3 was found to be the most overexpressed gene in the AUY922-resistant cells and we confirmed that this ITGB3 overexpression was associated with AUY922 resistance by western blotting (Fig. 2A). In addition, we found that ITGAv, as a representative counterpart of ITGB3, was also overexpressed in the AUY922-resistant cells (Fig. 2A). Because ITGB3 and ITGAv are well known to be regulated by miR-150 and miR-142, respectively [18–20], we confirmed that miR-150 and miR-142 were downregulated in our two AUY922 resistant NSCLC cell lines (Fig. 2B). Furthermore, FAK signaling was also found to be activated in these resistant cells, which was consistent with the well-known activation of FAK as a downstream pathway of ITGAvβ3 in various tumors (Fig. 2C) [21–23].
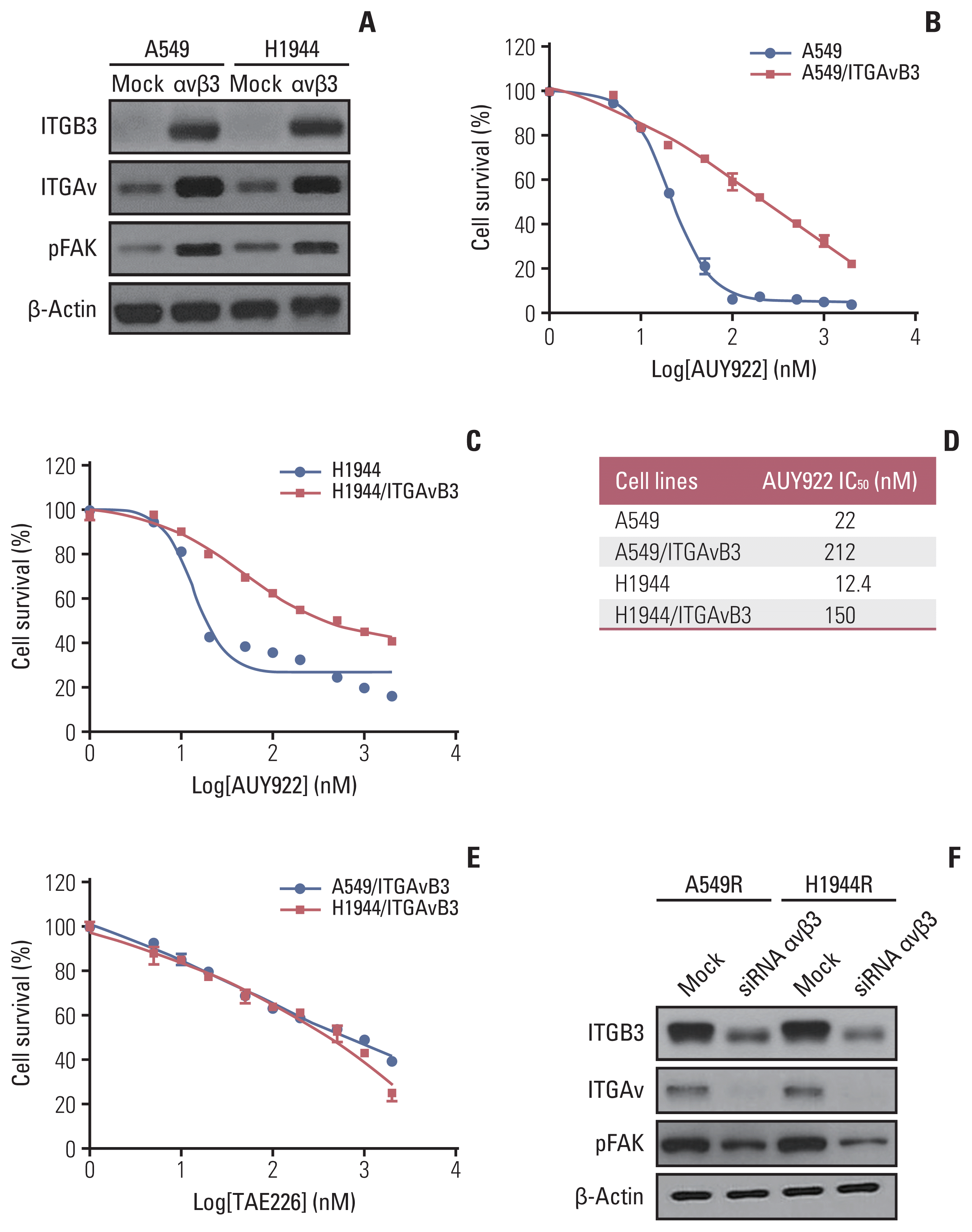
In our present study, we further investigated whether ITGAvβ3 can induce resistance to the HSP90 inhibitor, AUY922, in KRAS-mutant NSCLC cells. We hypothesized that ITGAvβ3 upregulation may induce this resistant phenotype through the activation of FAK signaling. To test this hypothesis, we first ectopically expressed ITGAv and ITGB3 by stable transfection in two integrin-negative parental KRAS-mutant NSCLC cells (Fig. 3A). Remarkably however, ITGAvβ3-transfected KRAS-mutant cells exhibited a higher IC50 to AUY922 than their mock-transfected counterparts (Fig. 3B–D). FAK activation in these stable cells (Fig. 3A) revealed that acquired AUY922 resistance might occur through FAK activation, as bypass (Fig. 2C), via overexpression of both ITGAv and ITGB3. In addition, we showed that ITGAvB3-induced AUY922-resistant cells were dose-dependently killed by FAK inhibitor (TAE226) (Fig. 3E). We also showed decrease of pFAK expression after knockdown of ITGAv and ITGB3, respectively, as well as integrin inhibitor treatment (Fig. 6E), in two AUY922 resistant cells (Fig. 3F).
2. AUY922-resistant KRAS-mutated NSCLCs show an EMT tendency via TSPAN7 induction
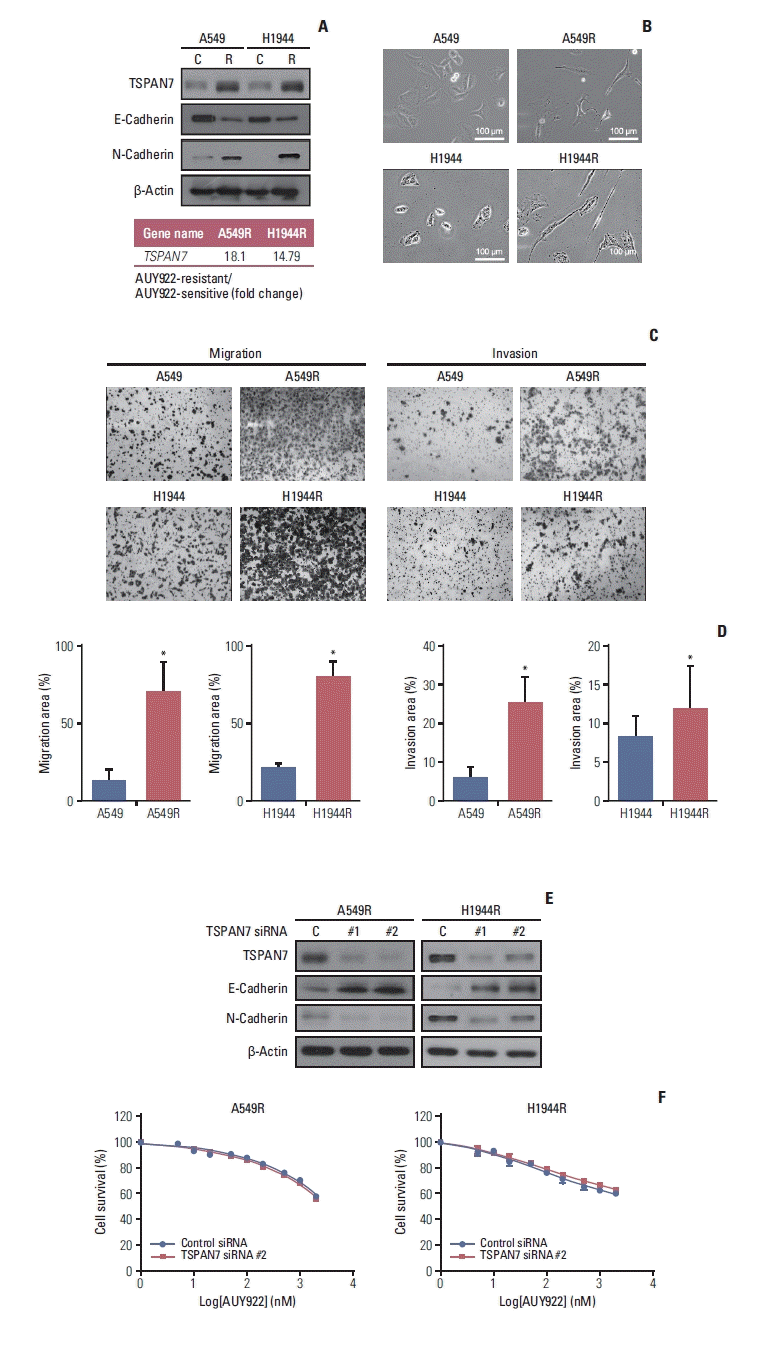
We selected another DEG, TSPAN7 (tetraspanin 7), from the comparison of our RNA sequencing data between AUY922-resistant and -sensitive cancer cells (Fig. 4A). TSPAN7-mediated EMT is well known as a key event in NSCLC migration and is regarded therefore a possible target for NSCLC therapy [24]. Although TSPAN7 has been implicated in the EMT of NSCLCs [24], the involvement of TSPAN7-mediated EMT in AUY922 resistance in KRAS-mutated NSCLCs has not been well explored. We thus examined the expression of two EMT markers (E-cadherin and N-cadherin) in our A549R and H1944R cells by western blotting analysis and observed an increased N-cadherin caused by a reduction in E-cadherin in the TSPAN7-induced resistant cells (Fig. 4A). In addition, an EMT-like morphology was evident in the resistant cells (Fig. 4B), as was augmented migration and invasion in Boyden chamber assays (Fig. 4C). To then examine whether TSPAN7 affects AUY922 resistance, we depleted this gene in the A549R and H1944R cells using two siRNAs. This TSPAN7 depletion led to significant N-cadherin overexpression (Fig. 4E). However, an CellTiter-Glo Luminescent Cell Viability Assay indicated that the TSPAN7-depleted A549R cells were still as resistant to AUY922 as the control siRNA-expressing cells (Fig. 4F).
3. The co-expression of ITGB3 and ITGAv caused by miR-150 and miR-142 downregulation induces AUY922 resistance
Previous studies have reported that TSPAN7 [24] and ITGB3 [20] are induced by microRNA-150 (miR 150) downregulation activity and that ITGAv is targeted of miR-142 [18,19,25]. Since both TSPAN7 and ITGB3 are co-direct targets of miR-150 [20,24], we examined whether this regulatory molecule controlled EMT and FAK activity through the blockade of TSPAN7 and ITGB3 expression, respectively. I ndeed, miR-150 expression in our two AUY922 resistant cells, A549R and H1944R, was 4-fold lower than in their parental A549 and H1944 counterparts (Fig. 2B).
Based on these results, we hypothesized that miR-150 and miR-142 may regulate FAK activation. To test this possibility, we investigated whether the restoration of miR-150 and miR-142 expression in AUY922 resistant cells (A549R and H1944R) would diminish FAK signaling. A549R and H1944R cells were transfected with miR-150 and miR-142, and its ectopic expression was confirmed by quantitative RT-PCR (Fig. 5A and E). As shown in Fig. 5B and F, ectopic miR-150 and miR-142 expression in A549R and H1944R cells led to a significant downregulation of phosphorylated FAK via the inhibition of ITGAvB3 formation through ITGB3 and ITGAv suppression, respectively. We next tested whether miR-150 and miR-142 could restore AUY922 sensitivity by inhibiting FAK signaling in A549R and H1944R cells. Indeed, ectopic miR-150 and miR-142 expression reversed the AUY922 resistance phenotype of these cells (Fig. 5C, D, G, and H). These results suggested that miR-150 and miR-142 could restore AUY922 sensitivity by blocking the FAK signaling bypass via suppression of ITGB3 and ITGAv expression, respectively.
4. ITGAvβ3-induced AUY922 resistance is FAK-dependent but is independent of the EMT mediated by TSPAN7
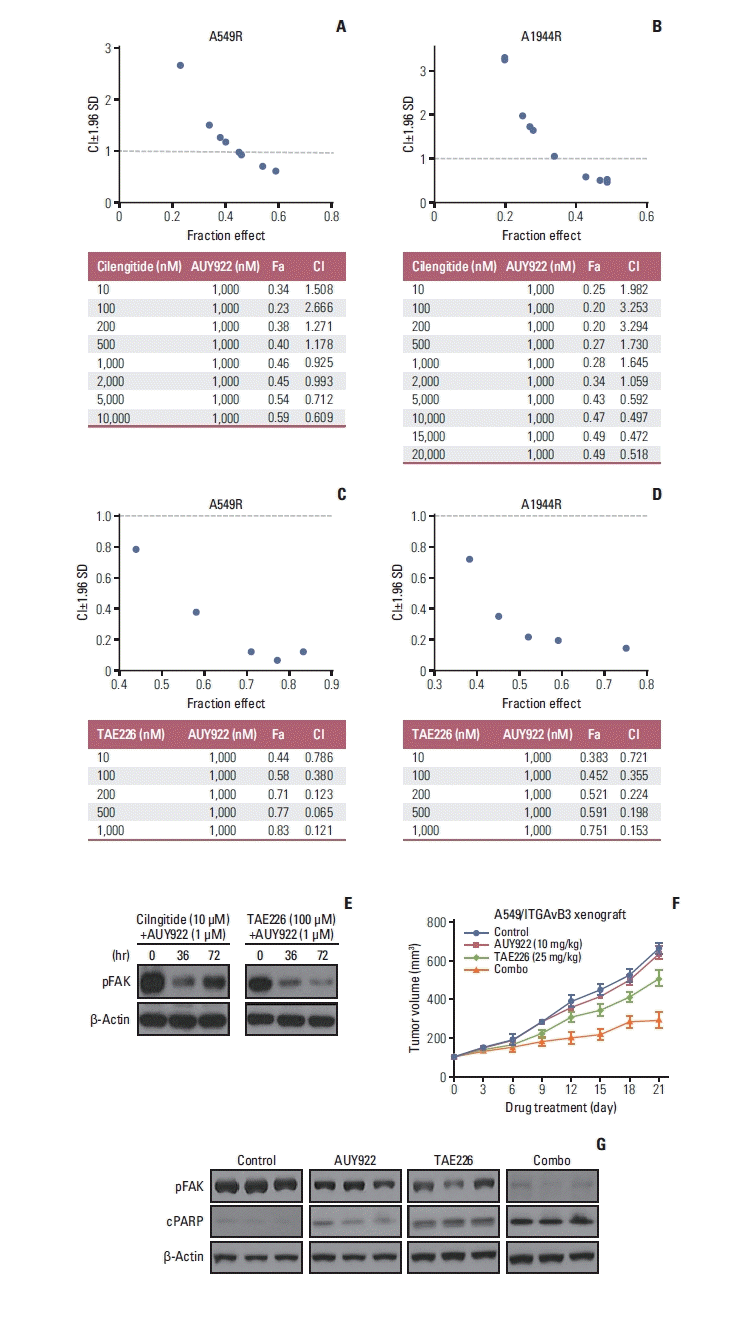
To test whether the combined inhibition of FAK and HSP90 could be a potent strategy for overcoming ITGAvβ3-mediated AUY922 resistance, we employed the Chou-Talalay method (61) using different concentrations of the ITGAvβ3 inhibitor, cilengitide, or the FAK inhibitor, TAE226, in combination with AUY922 in the AUY922-resistant cells (A549R and H1944R). TAE226 rendered A549R and H1944R cells sensitive to AUY922, with a significant degree of synergy (CI < 1) observed at all concentrations tested (Fig. 6C and D). Cilengitide also rendered A549R and H1944R cells sensitive to AUY922 as part of a high-dose combination treatment (Fig. 6A and B). In particular, in part of pFAK inhibition effect, the FAK inhibitor, TAE226 (100 nM), was found to be more effective than the ITGAvβ3 inhibitor, cilengitide (10 mM) in A549R cell (Fig. 6E). In addition, xenograft experiments with A549/ITGAvB3 cells revealed that a TAE226 and AUY922 combination reverted the ITGAvB3-induced AUY922 resistance and significantly attenuated tumor growth in comparison with either drug alone (Fig. 6F). FAK phosphorylation was less affected by AUY922 exposure in AUY922-resistant A549/ITGAvB3 xenografts (Fig. 6G), implying that the activation of FAK signaling may play a role in ITGAvB3-induced AUY922 resistance.
Discussion
During last decade, HSP90 inhibitors have been in the spotlight as treatments for other drug-resistant cancers, including KRAS-mutant tumors. At the early stage of the HSP90 inhibitor development, these agents showed potent effects against most tumor cells. However, a leading drug among this panel of inhibitors, AUY922, showed disappointing results in several clinical trials over the last 3 years and its development was discontinued as a single agent, even though several trials have continued to test it as part of a combination treatment regimen [9,15].
Biomarkers of first-generation HSP90 inhibitors, such as ganetespib, resistance have been suggested, including MCL1 [26] and JAK [27], but resistance biomarkers for second generation HSP90 inhibitors such as AUY922 are yet to be established. Significantly, we have here identified a novel candidate target that is related to AUY922. Our RNA sequencing analysis has indicated that ITGB3 shows a 29-fold increase in acquired AUY922 resistant cells compared to the parental controls. Integrins are well known as cell adhesion molecules that connect the cytoskeleton to the extra-cellular matrix or to other cells. These proteins consist of two noncovalently bound transmembrane α and β subunits. A blockade of integrin signaling has been demonstrated to be effective in inhibiting tumor growth, angiogenesis [28], and metastasis. Among the family members, ITGAvβ3 is highly expressed on activated endothelial cells as well as some tumor cells, but is not expressed in resting endothelial cells or most normal organs, suggesting its potential as a target for anti-cancer therapy. Prior studies have demonstrated that ITGAvβ3 induces EGFR–tyrosine kinase inhibitor resistance in KRAS-mutant NSCLC, in association with fibroblast growth factor-2, metalloproteinase matrix metalloproteinase 2, activated platelet-derived growth factor, insulin, and vascular endothelial growth factor receptors, thereby facilitating the optimal activation of cell proliferation, invasion and preventing apoptosis [28]. In addition, ITGAvβ3 has been demonstrated to induce cancer cell proliferation and survival via the activation of FAK [29].
ITGAv is one of the few integrin α subunits that associates with multiple β units, and each resulting complex belongs to the RGD ligand group. This integrin is regulated by miR-142 and miR-31, and there is evidence that it is also regulated by miR-100. We thus confirmed that both ITGAv and ITGB3 are overexpressed in the two AUY922 resistant cell lines analyzed in our present report (Fig. 2A), and established for the first time that stable ITGAvβ3 expression in cells directly induces AUY922 resistance. Interestingly, the two ITGAvβ3-overexpressing NSCLC cell lines we tested here not only showed AUY922 resistance but also indicated the involvement of FAK activation (Fig. 3), a well-known downstream pathway among several ITGAvβ3 signaling mechanisms. On the other hand, TSPAN7, another identified DEG in our current analyses, was found to be increased in our two AUY922 resistant cell lines. As TSPAN7 is known to promote the migration and proliferation of lung cancer cells via an EMT [24], we observed changes in the expression of EMT-related markers in AUY922-resistant cells i.e., overexpressed N-cad-herin and downregulated E-cadherin (Fig. 4A). When TSP-AN7 was knocked down by two different siRNAs, the E-cadherin level was restored in the AUY922-resistant cells (Fig. 4E). However, this EMT change caused by TSPAN7 did not affect the AUY922 sensitivity of the cells (Fig. 4F).
To elucidate how ITGAv and ITGB3 become overexpressed in AUY922-resistant cells, we explored the possible role of miRNAs which are known to regulate integrins [25]. ITGB3 is also known to be regulated by several different miRNAs. For example, miR-30 is significantly reduced when ITGB3 expression is significantly increased in breast tumor initiating cells [30]. miR-150 has also been shown to regulate megakaryocyte-erythrocyte progenitor cell development and the expression of ITGB3 on the surface of differentiated cells [30]. Moreover, miR-150 is expressed in various cancers, including cervical cancer and NSCLC [30,31] and promotes cell proliferation, migration, and invasion [32].
Our present results showed that ITGAv and ITGB3 are increased by miR-142 and miR-150, respectively (Fig. 2A and B). As miR-150 is reportedly an upstream regulator of EMT via TSPAN7 expression and contributes to ITGAvB3 complex formation via ITGB3 overexpression, we also showed that ITGAvβ3 overexpressing AUY922-resistant cells had downregulated miR-150 and miR-142 expression (Fig. 2B). In addition, the upregulation of miR-150 and miR-142 in our AUY922-resistant cell lines restored AUY922 sensitivity, which occurred through the suppression of FAK activation (Fig. 5) because ITGB3 and ITGAv could not be expressed, respectively. Accordingly, we assessed whether a combination of a FAK inhibitor (TAE226) or integrin inhibitor (cilengitide) with the AUY922 HSP90 inhibitor had a synergistic effect both in vitro and in vivo. The dual inhibition of FAK and HSP90 was a more potently synergistic combination treatment in the two AUY922 resistant cell lines and in xenograft tumors (Fig. 6A–D and F) and interestingly, FAK inhibition was maintained for longer by a dual blockade of FAK and HSP90 than of integrin and HSP90 (Fig. 6E). This FAK and HSP90 combination inhibitor treatment induced a greater apoptotic response, which may be more important in terms of killing effect in stable ITGAvB3 expressing cells (A549/ITGAvB3) (Fig. 6G).
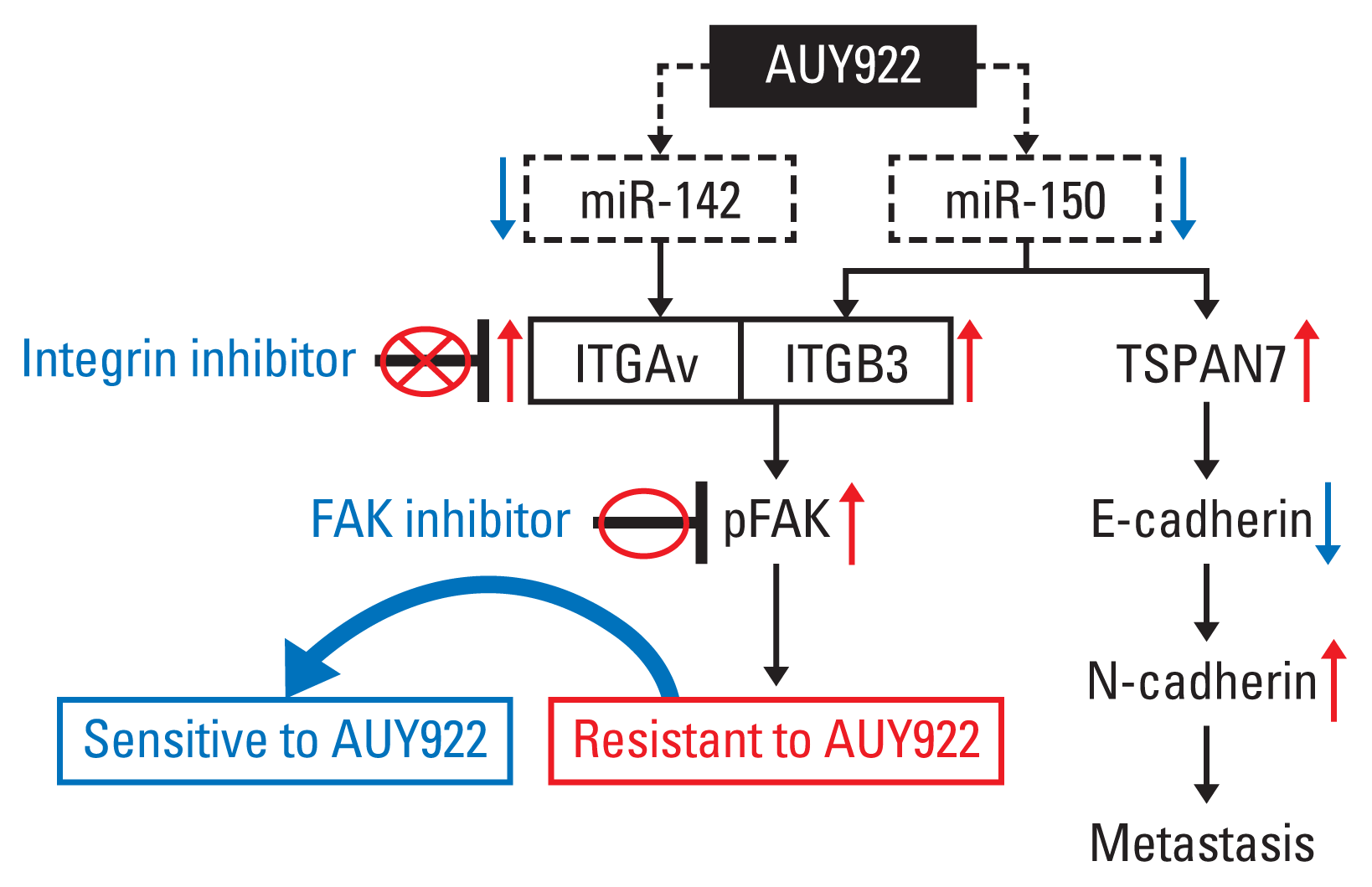
It was fortunate in our present analyses that we could check the mRNA levels of both ITGAv and ITGB3 in the tumor tissue obtained from a patient who had participated into a clinical trial of AUY922 and developed drug resistance. Although the sample was obtained from one patient only, we showed clinical correlation between ITGAvB3 expression and AUY922 resistance (S1 Fig.). Our present study findings suggest that the overexpression of ITGAv and ITGB3 is caused by miR-142 and miR-150 suppression, respectively. The induction of integrin AvB3 (ITGAvB3) will eventually result in AUY922 resistance via FAK activation, and therefore, the combination of a FAK inhibitor (TAE226), and not and integrin inhibitor, with a HSP90 inhibitor (AUY922) may be an effective strategy for overcoming ITGAvB3-induced acquired resistance (Fig. 7), or for preventing the occurrence of the resistance in KRAS-mutated tumors, or at least NSCLCs. We also suggest from these findings that next-generation HSP90 inhibitors could be more effective if they target both HSP90 and FAK.
 XML Download
XML Download