

 PDF
PDF Citation
Citation Print
Print


Introduction
Epidermal growth factor receptors (EGFR), a family of receptor tyrosine kinases, are well-known for their roles in tumorigenesis, such as cell proliferation, invasion, and angiogenesis [1]. Overexpression of and mutations in the EGFR are commonly found in non–small cell lung cancer (NSCLC), which lead to amplification of EGFR signaling and constitutive activation of EGFR [1]. EGFR–tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib, inhibit EGFR signaling cascades by competitive binding with ATP and result in the attenuation of tumor growth by inducing G0/G1 phase cell-cycle arrest [2]. Along with this principal effect, TKIs are also known to mediate autophagic flux in diverse cancer cells [3]. Moreover, targeting of autophagy with specific inhibitors leads to enhancement of gefitinib cytotoxicity in an EGFR-independent manner [4]. The elevation of autophagic flux in tumor cells is generally considered to play a cyto-protective role against anti-cancer drugs, and targeting the autophagy pathways appears to be a promising strategy to control tumor cell proliferation and acquired drug-resistance [5].
Autophagy is a catabolic degradative process in which cellular organelles are sequestered within double-membrane structures known as autophagosomes. Trafficking of auto-phagosomes to lysosomes catabolizes their cargo into amino acids, fatty acids, and carbohydrates, which promotes energy recycling and cell survival [6]. Although autophagy was first known as a non-selective catabolic process, the detection of autophagy receptors suggested a practical link between autophagy and ubiquitination [7]. Binding of agonists, including EGF, transforming growth factor-α, and amphiregulin, to the EGFR has been reported to mediate internalization and autophagic degradation of EGFR [8]. TKIs were also found to mediate EGFR degradation in cancer cells by accelerating ubiquitination and endocytosis of EGFR [9]. Furthermore, an EGFR harboring an L858R or delE746-A759 mutation shows increased sensitivity to TKI-induced degradation compared to wild-type (WT) or L858R/T790M mutant EGFR [9].
The delicate balance between ubiquitination via ubiquitination-related enzymes such as E3 ubiquitin ligase and deubiquitination via deubiquitinating enzymes (DUBs) is known to be highly associated with tumorigenesis [10]. Ubiquitin-specific peptidase 37 (USP37) is a DUB that removes ubiquitin from target proteins. Accumulating evidence indicates that deubiquitination by USP37 is essential for maintaining diverse biological processes in cancer cells. Deletion of USP37 results in mesenchymal epithelial transition by suppressing cell migration and invasion in breast cancer cells [11] and suppression of cell proliferation by hypoxia inducible factor α down-regulation [12]. Also, USP37 stabilizes Snail and c-Myc, which promote lung cancer cell migration [13,14]. Although USP37 appears as a potential therapeutic target to treat cancer cells, the molecular function of USP37 in the gefitinib-mediated autophagic process is still unclear.
MicroRNAs (miRNAs) are 22-nucleotide long, endogenously expressed non-coding molecules, which target specific mRNAs resulting in regulation of gene expression. Recently, microarray analysis of serum and blood samples from patients with hepatocellular carcinoma (HCC) revealed that miRNA levels, including miR-4484, miR-1281, and miR-4487, are markedly upregulated, in comparison to those in normal blood samples, and contribute to the diverse biological processes of cancer cells by targeting DUSP1, programmed death-ligand 1, and MUC1 [15]. Of note, miR-4487 was also identified to target Unc-51-like kinase 1 (ULK1) and regulate the ULK1-mediated autophagic process in neuroblastoma cells [16]. However, so far, very few studies address the physiological roles of miR-4487 in cellular processes.
Thus, in this study, we (1) examined the physiological roles of miR-4487 and USP37; (2) tested autophagic flux and whole-cell ubiquitination; and (3) studied the effects, if any, of miR-4487 on USP37 function in gefitinib-mediated EGFR degradation in NSCLC cell lines.
Go to : 
Materials and Methods
1. Cell culture and reagents
The human bronchial epithelial cell line, BEAS-2B; four NSCLC cell lines, PC-9, HCC827, H358, and H1650; and the gefitinib-resistant sub-cell lines, PC-9/GR and HCC827/GR were used in the study. All cell lines were gifted by Dr. Jin Kyung Rho in Asan Medical Center, Ulsan University (January 29, 2019). Each cell line was cultured in RPMI-1640 (HyClone, Logan, UT) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, Foster City, CA) at 37°C in 5% CO2. Each cell line was used within 20 passages after thawing each of the stock vials. Mycoplasma screening was performed every 4 months using endpoint PCR method (BioMycoX Mycoplasma PCR Detection Kit, Cellsafe, Yongin, Korea).
Mimic and inhibitor of miR-4487 (10 nM, Ambion, Foster City, CA) and siRNA for USP37 (5′-AGUCAUCAUUCCAAGAUACCUGACC-3′) (10 nM, Integrated DNA Technologies, Inc., Coralville, IA) were delivered into cells using the lipofectamine RNAiMAX reagent (Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. Gefitinib and the antibodies against poly(ADP-ribose) polymerase-1 (PARP), LC3B, integrin-α6, and β-actin were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Antibody against EGFR was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). Antibody against USP37 was obtained from the Proteintech Group, Inc. (Rosemont, IL). Bafilomycin A1 and 3-methyladenine (3-MA) were obtained from Sigma-Aldrich (St. Louis, MO).
2. Reverse transcription–PCR, and endpoint and quantitative PCR assay
Total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. cDNA was synthesized from 1 μg of total RNA using a PrimeScript RT reagent kit (TaKaRa Bio, Inc., Kusatsu, Shiga, Japan). Primers used for endpoint PCR were as follows: EGFR_F, 5′-CTCCAGGAAGCCTACGTGAT-3′ and R, 5′-GTCTTTGTGTTCCCGCCGGACAT-3′; GAPDH_F, 5′-AGGGCTGCTTTTAACTCTGGT-3′ and R, 5′-CCCCACTT-CATTTTGGAGGGA-3′. Expression level of miR-4487 was quantified using the TaqMan miRNA assay kit (Thermo Fisher Scientific, Foster City, CA) following the manufacturer’s protocol. The U6 small nuclear RNA was used as internal control. Relative quantitation was calculated using the comparative ΔΔCt method.
3. Western blot assay
Protein expression level was determined using western blot analysis following a standard protocol. In brief, cells were lysed with RIPA buffer (Invitrogen, Foster City, CA) containing proteases and a phosphatase inhibitor cocktail (Invitrogen). Whole cell lysates were loaded on sodium dodecyl sulfate polyacrylamide gel electrophoresis and separated by molecular weight using gel electrophoresis. After the transfer onto polyvinylidene difluoride membranes (0.2-μm pore size), the membrane was incubated with the primary antibodies, EGFR (1:1,000), LC3B (1:1,000), PARP (1:1,000), USP37 (1:1,000), integrin-α6 (1:1,000), and β-actin (1:1,000), and horseradish peroxidase–conjugated IgG was then used as the secondary antibody. Immunoreactive proteins were detected using AzureSpot 2.0 (Azure Biosystems, Inc., Dublin, CA).
To determine the expression level of EGFR on the plasma membrane, the plasma membrane was fractionized using the Plasma Membrane Protein Extraction Kit (Abcam, Cam-bridge, MA) following the manufacturer’s instruction. In brief, cells were homogenized and these homogenates were then centrifuged at 700 ×g for 10 minutes at 4°C. Supernatants were transferred to a new tube and centrifuged at 10,000 ×g for 30 minutes at 4°C. To purify plasma membrane proteins, pellets resuspended in upper phase buffer and low phase buffer were incubated on ice for 5 minutes, and then centrifuged at 1,000 ×g for 10 minutes at 4°C. The upper phase was carefully collected in a new tube and centrifuged at 10,000 ×g for 10 minutes at 4°C. The plasma membrane pellets were dissolved in 0.5% Triton X-100 in phosphate buffered saline (PBS) for the western blot assay.
4. Fluorescence-activated cell sorting analysis of cell cycle and apoptotic cell death
Cells were washed out with cold PBS and fixed with 70% ethanol at −20°C for 2 hours, and were then stained with 0.5 mL propidium iodide (PI)/RNase Staining Buffer (BD Biosciences, San Jose, CA) and PI mixture for 15 minutes at room temperature to determine cell cycle and apoptotic cell death. A minimum of 2×104 cells were analyzed using the FACScan analyzer (Becton Dickinson and Company, Franklin Lakes, NJ).
5. 3′-Untranslated region–reporter assay
WT or mutant fragments of 3′-untranslated region (3′-UTR) of USP37 were amplified and inserted downstream of the firefly luciferase reporter gene in the pEZX-FR02 vector (Genecopoeia, Rockville, MD). Next, 100 nM mimic of miR-4487 and 250 ng pEZX-EF02 vectors containing WT and mutated USP37 were co-transfected into HEK293 cells using lipofectamine RNAiMAX and 3000 reagents, respectively (Life Technologies Inc.). After an additional cell incubation of 48 hours, the luciferase activity (Fluc/Rluc) was assayed using the Luc-Pair Duo-Luciferase Assay Kits 2.0 (Genecopoeia).
6. Ubiquitination assay
The ubiquitination assay was performed using the UBI-QAPTURE-Q Kit (Enzo Life Sciences Inc., Farmingdale, NY) following the manufacturer’s protocol. In brief, whole cell lysates were collected and 250 μg/mL of proteins were mixed with UBI-QAPTURE-Q matrix and then washed out with HEPES buffer. The matrix was resuspended with HEPES buffer and then incubated overnight at 4°C. After incubation, ubiquitylated proteins with matrix were centrifuged at 5,000 ×g for 30 seconds and samples were prepared by adding them to 5× gel loading buffer for 5 minutes to quench. Each sample were boiled at 95°C for 10 minutes prior to use and used for western blot assay.
7. Cytotoxicity assay
Cytotoxicity was determined by measuring extracellular glucose-6-phosphate dehydrogenase (G6PD) using a Vybrant Cytotoxicity Assay kit (Invitrogen) following the manufacturer’s protocols. In brief, cells were seeded onto 96-well plates and incubated under indicated conditions. Cell culture medium devoid of cells was collected, and G6PD activity was determined at excitation and emission wavelengths of 544 and 590 nm, respectively. Results were expressed as the percentages of total G6PD detected in cell lysates from parallel wells.
8. Measurement of intracellular Ca2+
intracellular Ca2+ ([Ca2+]i) level was determined using a Ca2+-sensitive fluorescence dye, Fura-2/AM (Sigma-Aldrich). Cells loaded with Fura-2 (5 μM) were perfused with HEPES buffer containing 140 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1 mmol/L CaCl2, 10 mmol/L HEPES, and 10 mmol/L glucose (adjusted to pH 7.4 and 310 mOsm). Next, 1 mmol/L CaCl2 was replaced with 1 mmol/L EGTA in Ca2+-free HEPES buffer. After a brief washing out with HEPES buffer, Fura-2 fluorescence was monitored using 340 and 380 nm dual-wavelength excitation, and emission wavelength at 510 nm was collected using a sCMOS camera (Andor Technology Ltd., Belfast, UK). Collected data were analyzed using the MetaFluor Fluorescence Ratio Imaging software (Molecular Devices, San Jose, CA) and presented as the F340/F380 ratio.
9. Statistical analysis
Statistical analysis was conducted using the Origin software (OriginLab Corp., Northampton, MA). Data are presented as mean±standard deviation. of observations obtained from more than three independent experiments. Statistical differences were analyzed using one-way ANOVA followed by Tukey’s post-hoc test. Values of p < 0.05 were considered statistically significant.
Go to :
Results
1. Long-term treatment of NSCLC cells with gefitinib reduces endogenous level of EGFR and its localization on the plasma membrane
A growing body of evidence indicates that EGFR-TKIs not only inhibit EGFR activation by interfering with the binding of ATP, but also mediate EGFR degradation via ubiquitination and the autophagic process, both of which result in cell-cycle arrest and apoptosis [9,17,18]. We first examined the gefitinib-mediated EGFR degradation in diverse NSCLC cells, including the PC-9, PC-9/GR, HCC827, HCC827/GR, H358, and H1650 cell lines. Each cell was treated with gefitinib for the indicated time and the endogenous level of EGFR and cleavage of PARP and LC3B were evaluated. Fig. 1A and B show that long-term treatment of NSCLC cells, excluding PC-9/GR and HCC827/GR, with gefitinib (1 μM) in the absence of additional EGF treatment commonly resulted in decreased endogenous levels of EGFR in a time-dependent manner. Notably, PC-9 cells show a rapid decrease in EGFR level and an increase in PARP cleavage in response to gefitinib treatment; a significant decrease in EGFR levels in PC-9 (0.60468±0.12501) and HCC827 (0.46442±0.18503) cells was observed at 12 and 48 hours, respectively (Fig. 1A and B). Along with gefitinib-mediated EGFR decrease, the cleavage of LC3 in the PC-9 cells, but not in gefitinib-resistant cell lines, was found to increase in response to gefitinib treatment (Fig. 1A). To determine whether the decrease in EGFR levels was caused by reduction in EGFR transcription, we performed an RT-PCR analysis with total RNA of PC-9 cells treated with gefitinib in a time-dependent manner. Fig. 1C shows that the transcriptional activity of the EGFR gene did not change in response to gefitinib treatment. In addition, we found that the localization of EGFR on the plasma membrane appeared to decrease after 2 hours of gefitinib treatment (Fig. 1D). To further identify the gefitinib-mediated decrease in EGFR levels, we evaluated the effects of gefitinib on EGF-induced [Ca2+]i oscillations using a ratiometric assay. PC-9 cells cultured with or without gefitinib for different time periods were perfused with EGF-containing Ca2+-free HEPES buffer. Consistent with the decrease in EGFR on plasma membrane fraction, EGF-mediated [Ca2+]i oscillations were markedly abolished in 2 hours of gefitinib pretreatment cells. There results indicate that the long-term treatment of NSCLC cells with gefitinib induces internalization of EGFR and a decrease in its expression levels.
 | Fig. 1Effects of long-term treatment of gefitinib on non–small cell lung cancer cells. Different cell lines were incubated with gefitinib (1 μM) for the indicated time. (A) Endogenous levels of epidermal growth factor receptor (EGFR) and cleavage of poly(ADP-ribose) polymerase-1 (PARP) and LC3B were determined using a western blot assay in the PC-9 and PC-9/GR cell lines. (B) Endogenous level of EGFR in HCC827, HCC827/GR, H358, and H1650 cells. β-Actin levels were used as loading controls. Column graphs show relative fold change of each protein in gefitinib-treated cells compared with that in control (0; no-gefitinib). Data are mean±standard deviation. *p < 0.05 compared with control or between indicated group. (C) Reverse transcription polymerase chain reaction was performed to detect the transcriptional activity of EGFR in gefitinib-treated PC-9 cells. (D) PC-9 cells were treated with gefitinib (1 μM) for the indicated time, and total membrane was fractionized and isolated proteins from each fraction were used to detect EGFR levels. Integrin-α6 was used as loading control. PM, plasma membrane; TM, total membrane. (E) Cells were pretreated with gefitinib (1 μM) before the experiments, and PC-9 cells loaded with Fura-2AM were used to determine intracellular Ca2+ ([Ca2+]i) response to epidermal growth factor (EGF; 200 ng/mL) in the absence of extracellular Ca2+. Each trace represents [Ca2+]i mobilization in a single cell.
|
2. Gefitinib-mediated EGFR degradation is dependent on autophagic flux
Based on the knowledge that gefitinib mediates LC3B cleavage and EGFR degradation, we assumed that gefitinib-mediated EGFR degradation is dependent upon the auto-phagic flux. To further identify the link between the auto-phagic process and EGFR degradation, autophagy inhibitors, bafilomycin A1 (5 nM) and 3-MA (5 mM), were added to gefitinib-treated PC-9 and HCC827 cells for 1, 3, and 6 hours prior to the end of incubation. Fig. 2A and B show that the increase in EGFR degradation and LC3 cleavage by gefitinib is significantly restored by inhibition of autophagy with bafilomycin A1 and 3-MA. These data clearly indicate that gefitinib-mediated EGFR degradation is dependent on the autophagic process.
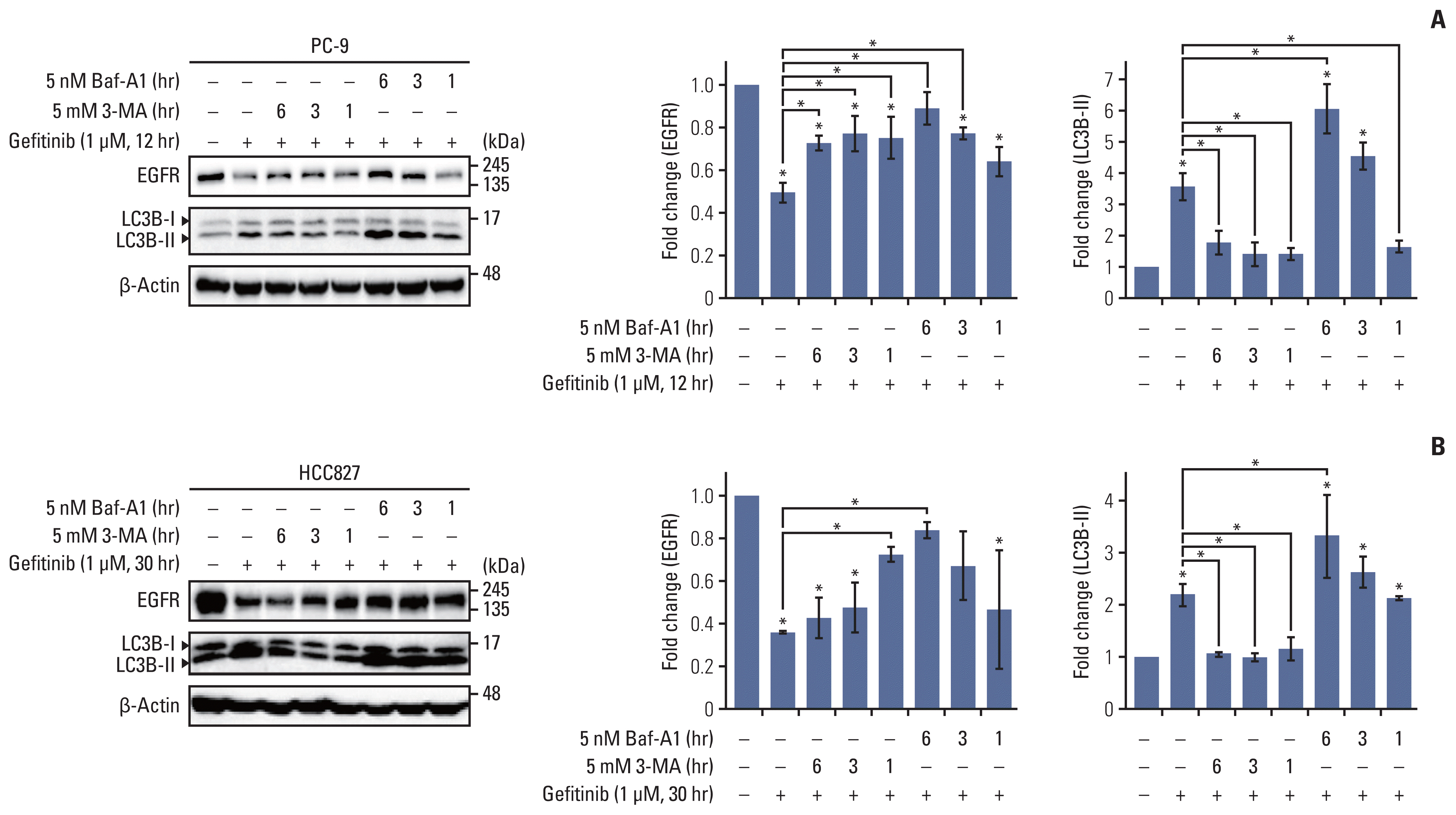 | Fig. 2Gefitinib-mediated epidermal growth factor receptor (EGFR) degradation is inhibited by autophagy inhibitors, 3-methyladenine (3-MA) and bafilomycin A1. Gefitinib (1μM)-exposed PC-9 (A) and HCC827 (B) cells were treated with bafilomycin A1 (5 nM) and 3-MA (5 mM) for 6 hours, 3 hours, and 1 hour before the end of incubation, and endogenous level of EGFR and LC3B cleavage were evaluated using western blot assay. Column graph data indicate fold change of each protein in inhibitor-treated cells compared with that in control (vehicle only). Data are represented as mean± standard deviation. * p < 0.05 compared with control or between indicated group.
|
3. Expression level of USP37 is negatively regulated by gefitinib in a time-dependent manner, and deletion of USP37 in gefitinib-treated cells synergistically enhances ubiquitination
With the discovery of autophagy receptors, ubiquitination is considered as a prerequisite for determining selective autophagy in eukaryotic cells [7]. We first screened the expression levels of USP37 and level of basal autophagic flux in gefitinib-sensitive and -insensitive NSCLC cells. Each NSCLC cell line was cultured under normal conditions, and whole cell lysates were harvested for western blot analysis. Our results show that USP37 expression was significantly down-regulated in NSCLC cells, excluding PC-9/GR cells, compared to that in the BEAS-2B cells. Contrary to the reduction of USP37 expression, LC3 cleavage in NSCLC cells was constitutively increased in PC-9, HCC827, H358, and PC-9/GR cells (Fig. 3A). Furthermore, endogenous USP37 in PC-9 cells was found to decrease on gefitinib treatment in a time-dependent manner (Fig. 3B). To further identify the role of USP37 in ubiquitination, siRNA of USP37 (siUSP37) was delivered into PC-9 cells, and cultured with or without gefitinib. Whole cell lysates were collected and used to determine the level of USP37 and ubiquitination. Fig. 3C indicates that gefitinib treatment and deletion of USP37 up-regulates ubiquitination and EGFR degradation. Moreover, deletion of USP37 and treatment of gefitinib was confirmed to increase LC3B cleavage respectively. Also, the use of gefitinib in siUSP37-deficient cells synergistically enhanced ubiquitination and EGFR degradation in PC-9 cells. These data demonstrate that USP37, a deubiquitinase, is down-regulated by gefitinib and results in synergistic effects on ubiquitination in cancer cells.
 | Fig. 3Endogenous level of ubiquitin-specific peptidase 37 (USP37) is down-regulated by gefitinib and USP37 depletion induces epidermal growth factor receptor (EGFR) degradation. (A) Endogenous levels of EGFR and LC3B cleavage were analyzed using western blot assay in BEAS-2B, PC-9, HCC827, H358, H1650, PC-9/GR, and HCC827/GR cell lines. Statistical data indicate fold change in USP37, LC3B cleavage in different cell lines compared with that in BEAS-2B cells. (B) Whole cell lysates isolated from gefitinib (1 μM)-treated PC-9 cells were used for western blot assay. Statistical data were presented as fold change of USP37 expression in cells (gefitinib-treated) compared to that in controls (no-gefitinib). Data are represented as mean± standard deviation. * p < 0.05 compared with control. (C) PC-9 cells were transfected with siUSP37 and then cultured with or without gefitinib (1 μM). After incubation, whole cell lysates were used for determining the endogenous levels of USP37, EGFR, LC3B cleavage, and ubiquitination.
|
4. miR-4487 is upregulated by gefitinib and directly targets USP37 to enhance gefitinib-mediated ubiquitination
Next, we investigated the upstream modulators of gefitinib-mediated USP37 down-regulation. Recently, it has been found that miR-4487 functions as a regulator in the cellular processes of cancer cells by targeting diverse genes [15,16]. Differential expression of miR-4487 in NSCLC cells was identified using the TaqMan primer for miR-4487. Fig. 4A shows that each NSCLC cell line strongly expresses miR-4487 compared to the expression in BEAS-2B cells. Moreover, the level of miR-4487 in PC-9 cells increases in response to gefitinib (Fig. 4B). To investigate the role of miR-4487 in gefitinib-mediated EGFR degradation and ubiquitination, transfection efficiency and specificity between miR-4487 and USP37 were validated using real-time quantitative PCR (qPCR) and 3′-UTR reporter assay, respectively. The left panel of Fig. 4C indicates that the level of miR-4487 was markedly increased in miR-4487-mimic-transfected cells. Furthermore, the 3′-UTR reporter assay revealed that miR-4487 directly interacts with USP37 and significantly suppresses USP37 expression (right panel of Fig. 4C). We also determined the effects of miR-4487 on the endogenous expression of USP37 and EGFR. Mimic and inhibitor of miR-4487 were transfected into each NSCLC cell line, to evaluate the level of endogenous USP37. Fig. 4D shows that the introduction of miR-4487 mimic in the NSCLC cell lines led to a significant reduction in USP37 levels in all cell lines, except HCC827, compared to those in the control. Also, Fig. 4E demonstrates that the introduction of mimic of miR-4487 in PC-9 cells mediated a marked reduction in EGFR levels, increased LC3 cleavage, and synergistically enhanced gefitinib-mediated EGFR degradation and LC3 cleavage. As shown in Fig. 4F, mimic of miR-4487 synergistically enhanced ubiquitination in PC-9 cells. These findings demonstrate that miR-4487 directly targets USP37, and that the suppression of USP37 by miR-4487 results in enhanced EGFR degradation and ubiquitination.
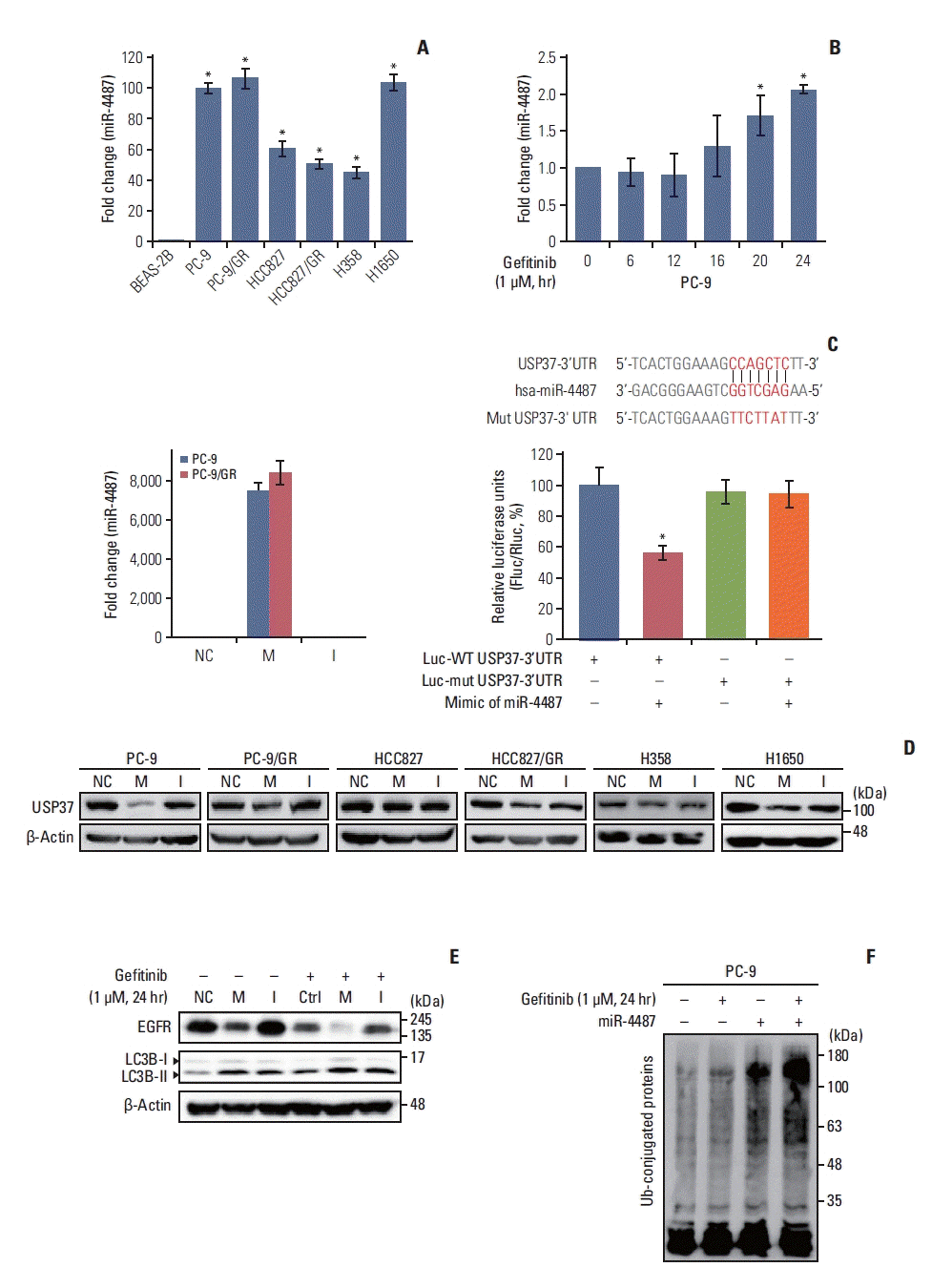 | Fig. 4miR-4487 directly targets ubiquitin-specific peptidase 37 (USP37) and promotes epidermal growth factor receptor (EGFR) degradation and ubiquitination. (A) Differential expression of endogenous miR-4487 was evaluated in BEAS-2B and non–small cell lung cancer (NSCLC) cells using TaqMan primers. Statistical data were presented as fold change of miR-4487 levels in the tested cell lines compared to that in BEAS-2B (control) cells. Data are represented as mean±standard deviation (SD). *p < 0.05 compared with BEAS-2B cells. (B) Endogenous level of miR-4487 was measured in gefitinib (1 μM)-treated PC-9 cells using TaqMan primers. Statistical data were presented as fold change in miR-4487 levels in gefitinib-treated cells compared to that in control (0; no-gefitinib). Data are represented as mean±SD. *p < 0.05 compared with non–gefitinib-treated cells. (C) PC-9 and PC-9/GR cells were transfected with miR-4487 mimic and inhibitor. Total RNA was used for quantitative polymerase chain reaction analysis (left panel). The wild-type (WT) and mutant sequences of USP37 3′-untranslated region (3′-UTR) are listed for comparison. HEK293 cells were co-transfected with either WT or mutated 3′-UTR plasmid and with miR-4487 mimic. Luciferase activity was measured at 48 hours after transfection. Firefly luciferase activity was normalized to Renilla luciferase activity and presented as relative to control (Luc-WT USP37 3′-UTR w/o miR-4487). Data are represented as mean±SD. *p < 0.05 compared to control. I, miR-4487 inhibi tor; M, miR-4487 mimic; NC, negative control. (D) miR-4487 mimic and inhibitor was transfected into NSCLC cell lines and endogenous USP37 expression was determined using the western blot assay. (E) After transfection with NC, M, and I, PC-9 cells were exposed to gefitinib (1 μM) for 24 hours. The endogenous levels of EGFR and LC3B cleavage were analyzed using a western blot assay. (F) miR-4487 mimic was transfected into PC-9 cells and then exposed to gefitinib (1 μM) for 24 hours. Whole-cell ubiquitination was assessed using the UBI-QAPTURE-Q Kit.
|
5. Introduction of miR-4487 and siUSP37 in PC-9 cells enhances cytotoxicity and apoptotic cell death
One remaining question was whether miR-4487/USP37-regulated EGFR degradation is associated with cytotoxicity and apoptotic cell death. Elevated autophagic flux in response to EGFR-TKIs has been implicated in the cytoprotective process rather than in inducing apoptotic cell death [5]. To gain an insight into the miR-4487/USP37-modulated EGFR degradation in determining cell fate, we examined the effects of miR-4487 and USP37 on apoptotic cell death and cell-cycle arrest. PC-9 cells were incubated under the indicated conditions, and PARP cleavage and G6PD release were evaluated. Fig. 5A and B demonstrate that gefitinib treatment in miR-4487 mimic and siUSP37-transfected PC-9 cells significantly enhances PARP cleavage and G6PD release compared to that in gefitinib-treated non-transfected cells. Also, overexpression of the miR-4487 inhibitor suppressed gefitinib-mediated PARP cleavage and G6PD release compared to that in only-gefitinib-treated cells (Fig. 5A and B). To further identify the roles of miR-4487 and USP37 on gefitinib-mediated cell-cycle arrest, we performed the fluorescence-activated cell sorting analysis. The 24-hour incubation of gefitinib with PC-9 cells markedly increased the cell population of G0/G1 phase by ~17% compared to that in negative control, whereas the population of apoptotic cells in the sub-G0/G1 phase remained unchanged (Fig. 5C). Interestingly, overexpression of miR-4487 (5.432%±1.030%) and deletion of USP37 (8.494±0.501) led to a significant increase in gefitinib-mediated apoptotic cell death in miR-4487 mimic and siUSP37-transfected cells compared to that in only-gefitinib-treated cells (1.766±0.336). These results suggest that the up-regulation of ubiquitination and autophagy by miR-4487/USP37 enhances gefitinib-mediated cytotoxicity and apoptotic cell death.
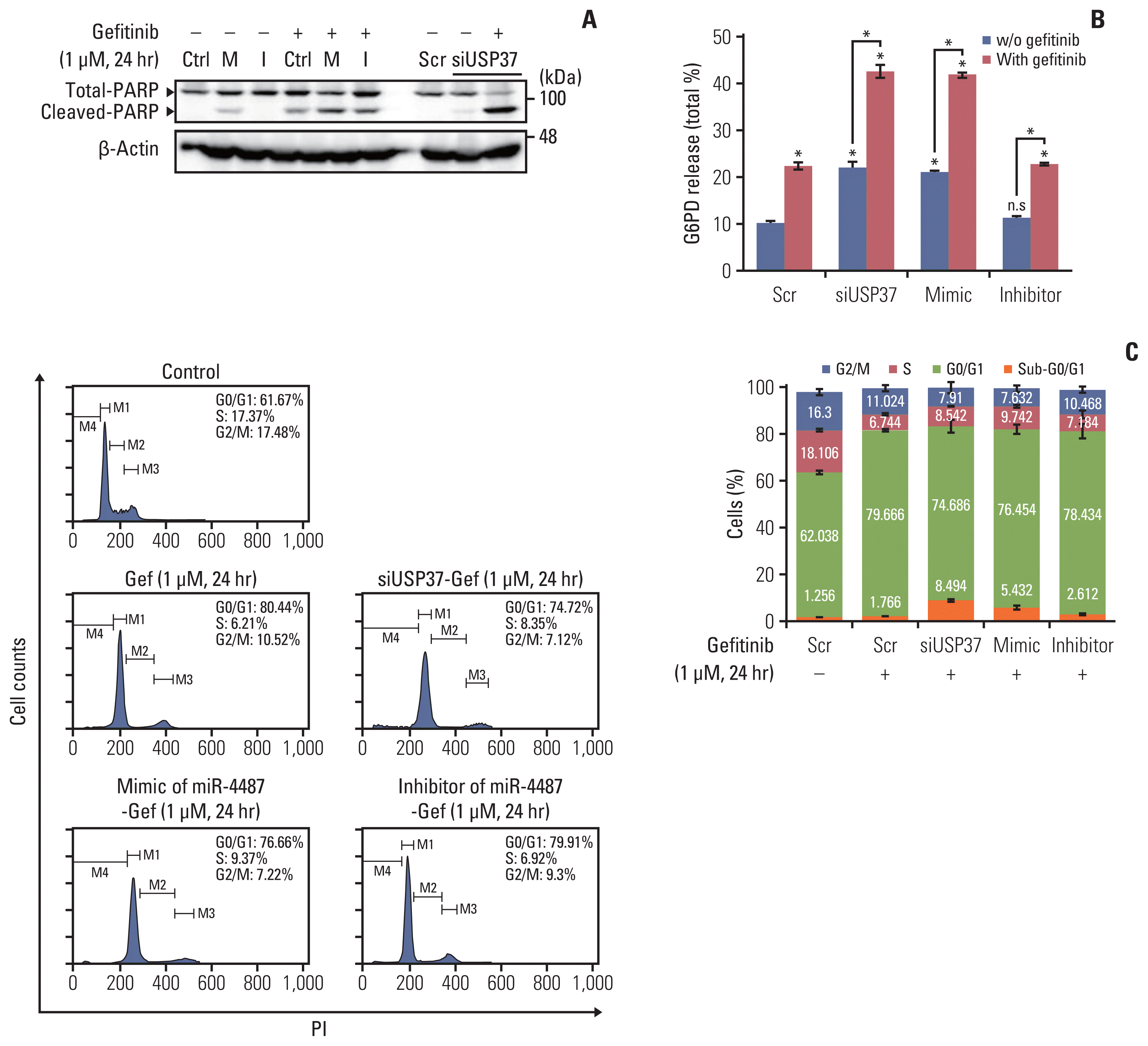 | Fig. 5Effects of miR-4487 overexpression and ubiquitin-specific peptidase 37 (USP37) depletion on gefitinib-mediated poly(ADP-ribose) polymerase-1 (PARP) cleavage, cytotoxicity, and apoptotic cell death. PC-9 cells were transfected with miR-4487 mimic (M)/inhibitor (I) and siUSP37 for 48 hours. Next, the cells were incubated for 24 hours with gefitinib (1 μM) and used to determine PARP cleavage (A), cytotoxicity (B), and cell cycle (C). (B) Cytotoxicity was evaluated by measuring glucose-6-phosphate dehydrogenase (G6PD) release. Data are presented as proportion of released G6PD (% total) and calculated as the mean±standard deviation. *p < 0.05 compared with scramble or between indicated groups. (C) Cells (2×104) stained with propidium iodide/RNase buffer were evaluated using flow cytometry. Data are represented as percentages and compared to those in control (scramble without gefitinib).
|
Go to :
Discussion
Ligands binding to EGFR in NSCLC cells consequently mediate EGFR dimerization and auto-phosphorylation in the carboxyl-terminus of EGFR to relay the diverse signaling pathways [19]. Small molecule EGFR-TKIs, such as gefitinib and erlotinib, suppress auto-phosphorylation of EGFR by competing with ATP, and, thus, inactive EGFR is internalized and endocytosed [8]. Endocytic trafficking of EGFR is known to be dependent on the type of ligands and mutations in the EGFR kinase domain [8,20,21]. Of note, Citri et al. [9] reported that TKIs also mediate degradation of ErbB-1 and -2 molecules by enhancing ubiquitination and endocytosis apart from their inhibitory effects on tyrosine phosphorylation [9]. In the present study, we found that long-term treatment with gefitinib in NSCLC cells differentially induces EGFR degradation depending upon the mutations in EGFR. PC-9 and HCC827 cells harboring the delE746-A750 mutation showed a marked reduction in the EGFR level, whereas gefitinib-resistant sub-cell lines, PC-9/GR and HCC827/GR, did not (Fig. 1). Also, H358 cells harboring WT EGFR showed no reduction in EGFR levels until 72 hours of gefitinib treatment. Subsequent results showed that gefitinib has no effect on the transcriptional activity of EGFR, but caused rapid reduction of the surface expression of EGFR and EGF-induced Ca2+ oscillations (Fig. 1). Ray et al. [22] also showed that EGFR degradation is differentially regulated by erlotinib depending upon the mutations harbored in EGFR. Also, erlotinib is found to induce polyubiquitination and proteasomal degradation in a c-Cbl–independent manner, and erlotinib-mediated EGFR degradation enhances the efficacy of radiation therapy and chemotherapy [22]. Consistent with these reports, our findings also demonstrated that prolonged treatment of NSCLC cells with gefitinib elicits an increase in ubiquitination and autophagic influx, which contributes to EGFR degradation (Figs. 2 and 3).
Our results suggest the synergistic effects of gefitinib treatment and suppression of USP37 expression on whole-cell ubiquitination. Furthermore, we proved that gefitinib up-regulates the miR-4487 level, which directly targets USP37 and results in the enhancement of the sensitivity to gefitinib (Figs. 3–5). To our knowledge, the present study provides the first evidence of the direct interaction between miR-4487 and USP37, and the synergistic effects of the genetic manipulation of miR-4487 on gefitinib-induced cell-cycle arrest and apoptotic cell death. Although previous microarray analyses have indicated the differential expression of miR-4487 in cancer serum samples [15,23], only a few studies have reported the biological roles of miR-4487 in specific cancer cellular processes. Down-regulated expression of miR-4487 by Aβ25–35 in SH-SY5Y cells is associated with Aβ-mediated pathophysiology, and overexpression of miR-4487 resulted in inhibition of apoptosis [24]. According to Chen et al. [16], miR-4487 plays an inhibitory role in ULK1-mediated autophagy in SH-SY5Y cells by targeting ULK1. In contrast, we found that miR-4487 exhibited a positive effect on gefitinib-mediated ubiquitination and autophagy. Apart from providing evidence of the direct targeting of USP37 by miR-4487, our results also show that the prolonged treatment of NSCLC cells with gefitinib resulted in an increase in miR-4487 and a decrease in USP37 levels, which led to the induction of whole-cell ubiquitination and autophagic flux (Figs. 3 and 4). In comparison to the previous report by Chen et al. [16], the positive role of miR-4487 in the present study is controversial. Although the controversial roles of miR-4487 in the autophagy process are not clearly explained in the present study, we assumed that miR-4487 may interact with different targets depending on the autophagy-inducing conditions. Our findings show that elevated miR-4487 in NSCLC cells under normal conditions enhances cellular ubiquitination and autophagy by targeting USP37.
One noteworthy finding in the present study is that the deficiency of USP37 in NSCLC cells by the overexpression of either miR-4487 or siUSP37 significantly increases not only autophagic flux, but also apoptotic cell death (Fig. 5). Elevation of autophagic flux in EGFR-TKI-treated cancer cells has been widely considered to have cytoprotective roles, and inhibiting autophagy can be a useful tool for overcoming drug-resistance [5,25]. However, the physiological role of autophagy in cancer cells is still unclear. As autophagy receptors, such as p62/SQSTM1 and NBR1, have been identified, the relationship between ubiquitination and selective autophagy is evident [26,27]. Our pharmacological study demonstrated that gefitinib-mediated EGFR degradation is dependent on the intracellular autophagic flux (Fig. 2). Moreover, elevation of autophagic flux by deletion of USP37 and overexpression of miR-4487 is closely associated with level of ubiquitination (Figs. 3 and 4). Interestingly, the expression levels of USP37 were shown to vary with cell origin and tissue types. Dobson et al. [28] found that USP37 plays a tumor-suppressive role in neuronal cancers, and was down-regulated in human medulloblastoma cells. In contrast, the level of USP37 in breast cancer tissues was highly upregulated and it participated in diverse cancer cell processes [11]. Our findings clearly show low levels of USP37 in NSCLC cells, which has synergistic effects on gefitinib-mediated ubiquitination and apoptotic cell death (Figs. 2 and 5). Taken together, our results show that the prolonged treatment of NSCLC cells with gefitinib mediates strong degradation of EGFR, elevating intracellular ubiquitination and autophagy and resulting in apoptotic cell death.
In summary, the present study demonstrates that gefitinib-mediated EGFR degradation is dependent on the mutation variations in the EGFR kinase domain, and miR-4487 contributes to the subsequent elevation of gefitinib-mediated ubiquitination, autophagy, and apoptotic cell death by directly targeting USP37. These results suggest a ‘non-classical’ effect of gefitinib on NSCLC cells, and miR-4487 emerges as a strong target for treatment in NSCLC patients.
Go to :
 XML Download
XML Download