

 PDF
PDF Citation
Citation Print
Print


Introduction
Immunotherapy plays an important role in the treatment of malignant tumors. For advanced solid malignant tumors that cannot be directly surgically resected, immunotherapy is a potential alternative to radiotherapy and chemotherapy [1,2]. Programmed cell death protein 1 (PD-1), considered a crucial target in tumor immunotherapy, is expressed at elevated levels on the membrane surfaces of activated T cells. As the ligand of PD-1, programmed death-ligand 1 (PD-L1) is highly expressed on the surface of certain malignant tumor cells. The interaction between PD-1 and PD-L1 render T cells unresponsive or ‘exhausted’ [3], which can result in reduced secretion of cytokines, such as interleukin 2 (IL-2), and inhibition of T cell proliferation [4]. Using immune checkpoint inhibitors to block the interaction between PD-1 and PD-L1 reverses the unresponsiveness or ‘exhaustion’ of T cells and strengthens antitumor immunity levels [5]. Some anti–PD-1 monoclonal antibodies, such as nivolumab and pembrolizumab, have been approved by the Food and Drug Administration for the treatment of certain cancers in which PD-L1 is highly expressed, such as unresectable melanoma and non-small cell lung cancer [6–9].
However, due to limitations associated with disease control rates and overall response rates in human clinical trials, as well as the cost and production of monoclonal antibodies, there has been an increase in the development of complementary approaches that are based on peptides [10]. Compared with those of monoclonal antibodies, the synthesis and quality control of peptides are simple; additionally, peptides rarely induce immunogenicity, can penetrate deep into tissues, and cost less than monoclonal antibodies to produce [11,12].
In our previous studies, we identified a peptide that binds to PD-1 (nABP284) through phage display technology. However, the weak affinity (11.8 μM) of this peptide for PD-1 indicated that there is great opportunity for optimization. We improved nABP284 as follows: (1) we analyzed the sequence homology of nABP284 and the extracellular domain of PD-L1; (2) we predicted the key region in which nABP284 binds to PD-1; (3) we searched for the hot spot in the corresponding region in the extracellular domain of PD-1 [13,14]; (4) we predicted the potential amino acids that may bind to the hot spot in the extracellular domain of PD-1 and performed computer-simulated structural analysis by using AutoDock and PyMOL [15,16]; and finally, (5) we obtained nABPD1 by optimizing nABP284.
Improving cytokine-induced killer (ICIK) cells were transformed from human peripheral blood monocytes (PBMCs) by incubation with cytokines. The efficacy and feasibility of ICIK cell treatment has been demonstrated by clinical trials [17,18]. Our previous studies have also shown that ICIK cell therapy can restore immune function and prolong survival in patients with head and neck squamous cell carcinoma [19,20].
In our study, compared with nABP284, nABPD1 had a significantly enhanced affinity for PD-1. The experiments described below systematically demonstrate that nABPD1 can specifically bind to PD-1, can block the interaction between PD-1 and PD-L1 and shows better efficacy than nABP-284. Our results show that nABPD1 can more effectively block the binding of human recombinant PD-L1 protein to PD-1 in neutralization experiments. nABPD1 also shows better effects than nABP284 in terms of reversing the inhibition of Jurkat T cells and ICIK cells mediated by Cal27 cells expressing high levels of PD-L1.
Our experiments demonstrate an efficacious approach for optimizing PD-1–binding peptide and obtaining a new peptide with high affinity for PD-1 (nABPD1). This study provides a basis for further in vivo experiments and the exploitation of anti–PD-1 peptides.
Materials and Methods
1. Cell isolation and culture
The human acute T lymphocytic leukemia cell line (Jurkat T cells) and the human tongue squamous cell carcinoma cell line (Cal27 cells) were purchased from the cell bank of the typical Culture Preservation Committee of the Chinese Academy of Science (Shanghai, China). The Jurkat cells were cultured in RPMI 1640 (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (04-001-1ACS, Biological Industries, Kibbutz Beit-Haemek, Israel). The Cal27 cells were grown in Dulbecco’s modified Eagle’s medium/F12 (Sigma-Aldrich) supplemented with 10% fetal bovine serum (10099-141, Gibco, Grand Island, NY). PBMCs were isolated from peripheral blood donated by healthy adults through density gradient centrifugation and cultured in RPMI 1640 (Sigma-Aldrich) supplemented with 10% fetal bovine serum (10099-141, Gibco), interferon γ (IFN-γ; 300-02, 2000 U/mL, PeproTech, Rocky Hill, NJ), and IL-2 (200-02, 10 ng/mL, PeproTech).
2. Design and synthesis of the new peptide
We searched for the key regions through which human PD-L1 binds to human PD-1 and compared the amino acid sequences in the key regions of PD-L1 with the sequence of nABP284, which was identified by phage display technology in our previous study. The 2RLKEIA7 motif in nABP284 (SRLKEIANSPTQFWRMVARNTLGNGAKQSLNIEHARL) was considered the key region for specific binding. Based on the cleft between Y68 and E136 of PD-1 near the key binding region, which was just enough to accommodate the circular structure on the amino acid side chains, we hypothesized that adding histidine residues could enhance the binding affinity of nABP284 for PD-1 considering that histidine has an imidazole ring; this hypothesis was confirmed by AutoDock and PyMOL simulations. Thus, we added branched peptide (SHHHRL) to the N-terminus of nABP284 to obtain nABPD1. The new peptide was synthesized by China Peptide Company (Jiangsu, China) through solid-phase synthesis, and electrospray ionization–mass spectrometry/high-performance liquid chromatography (ESI-MS/HPLC) confirmed the sequence of the new peptide and its purity.
3. Surface plasmon resonance analysis of peptides
We used the Biacore T100 platform (GE Healthcare, Pittsburgh, PA) to measure the binding affinity of peptides through surface plasmon resonance (SPR). The surface of the chip (CM5) was esterified using the crosslinking agent EDC/NHS at pH 4.5, human recombinant PD-1 protein (PD1-H5221, Acrobiosystems, Newark, NJ) was conjugated to the chip (CM5) at a concentration of 5 μg/mL in coupling buffer (10 mM sodium acetate, pH 4.5), and excess active carboxyl groups on the surface of the CM5 chip were blocked with ethanol hydrochloride (pH 8.5). The channel that did not bind the human recombinant PD-1 protein was set as the reference. All the SPR signals were calibrated by subtraction of the reference channel response. The peptides were diluted in running buffer (pH 7.4, 100 mM Tris, 150 nM NaCl, 0.005% Tween-20). We first conducted the experiment with 1.25 μM (nABP284) and 0.125 μM (nABPD1), and we chose the concentration gradient of peptides according to the response values. For nABP284 analysis, seven concentrations (20, 10, 5, 2.5, 1.25, 0.625, and 0.3125 μM) were used for a round of SPR measurements; similarly, for nABPD1, seven concentrations (2, 1, 0.5, 0.25, 0.125, 0.0625, and 0.03125 μM) were used for a round of SPR measurements. The CM5 chip was regenerated with 10 mM glycine-HCl (pH 2.5) after each round of association and dissociation. The affinity (KD value) was calculated by fitting the peptide binding curve on the blank subtracted sensorgrams (Biacore Evaluation Software, Cytiva, Marlborough, MA).
4. Immunofluorescence and flow cytometry analysis of binding specificity
In the following experiments, an experimental group and a control group were included. For the experimental group, phorbol 12-myristate 13-acetate (PMA; 1652981, 50 ng/mL, PeproTech) and ionomycin calcium salt (ionomycin, 5608212, 1 μg/mL, PeproTech) were used to stimulate the expression of PD-1 in Jurkat T cells for 24 hours, while unstimulated Jurkat T cells expressing low levels of PD-1 served as the control group. For immunofluorescence analysis, Jurkat T cells (1×106) were harvested and blocked with 2% bovine serum albumin (BSA; Sigma-Aldrich) at room temperature (RT) for 1 hour to reduce nonspecific binding. After blocking, the cells were collected and incubated with 10 μM fluorescein isothiocyanate (FITC)–conjugated PD-1–binding peptides at 37°C for 1 hour. Then, the cells were washed with phosphate buffered saline (PBS) three times and fixed with 4% paraformaldehyde. After three washes with PBS, the cells were incubated with an anti–PD-1 antibody (ab52587, 1:100, Abcam, Cambridge, MA) overnight at 4°C. After three washes with PBS, a goat polyclonal secondary antibody against mouse IgG (ab150115, 1:500, Abcam) was allowed to bind to the anti–PD-1 antibody for 1 hour at 4°C. After three washes with PBS, the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; 1:1,000, Sigma-Aldrich), and the cells were mounted with fluorescence mounting medium (S3023, DAKO, Carpinteria, CA). Furthermore, a competition assay was performed using confocal microscopy, and the images were analyzed by ImageJ (National Institutes of Health, Bethesda, MD). For flow cytometry analysis, cells (2×105) were harvested and blocked with 2% BSA (Sigma-Aldrich) for 1 hour at RT and then incubated with 10 or 40 μM FITC-conjugated PD-1–binding peptides for 1 hour at 4°C. An anti–PD-1 antibody was used as a positive control. After blocking, the cells were incubated with a PD-1 antibody (130-120-389, 1:50, Miltenyi Biotec, Bergisch Gladbach, Germany) for 10 minutes at 4°C. After three washes with PBS, the cells were resuspended in PBS and analyzed with a flow cytometer (CytoFLEX, Beckman Coulter, Brea, CA).
5. Neutralization experiment with peptides
Jurkat T cells were divided into two groups. For the experimental group, cells were pre-stimulated with PMA (1652981, 50 ng/mL, PeproTech) and ionomycin calcium salt (ionomycin, 5608212, 1 μg/mL, PeproTech) for 24 hours, and unstimulated Jurkat T cells were used as the control group. The cells (5×105) were harvested and washed once with fluorescence-activated cell sorting (FACS) buffer (PBS containing 2% BSA). Biotinylated human PD-L1 (PD1-H82F3, Acro Biosystems) was diluted with FACS buffer to a concentration of 4 μg/mL. PD-1–binding peptides were diluted to various concentrations (0.8, 2.66, 8, 26.6, and 80 μM) with FACS buffer. Then, two working solutions (PD-L1 protein diluent and peptide diluent) were mixed well in equal volumes, and 100 μL of each mixture was added to the tube with the cell pellet and incubated at 4°C for 1 hour. The cells were washed with FACS buffer three times, and APC streptavidin diluent (405207, 0.12 μg/mL, BioLegend, San Diego, CA) was incubated with the cells at 4°C for 1 hour. The cells were washed with FACS buffer three times, resuspended in 500 μL of PBS, transferred to a flow tube and analyzed by flow cytometry (CytoFLEX, Beckman Coulter).
6. Cytotoxicity assay
Cytotoxicity assays were performed using a Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan). Jurkat T cells (4,000 cells per well), ICIK cells (4,000 cells per well), or Cal27 cells (2,000 cells per well) were seeded in 96-well plates and cultured for 24 hours. Various concentrations of each peptide (1, 2, 4, 8, 16, 32, and 64 μM) were added to the culture medium. The cells were cultured in a cell incubator for 24 hours, after which 10 μL of CCK-8 solution was added to each well, and the mixture was cultured in a cell incubator for another 2 hours. A Victor X5 Multilabel Plate Reader (PerkinElmer, Singapore) was used to measure the absorbances of each well at 450 nM. Cell viability (%) was calculated using the following formula: cell viability (%)=[(Asample–Abaseline)/(Acontrol–Ablank)]×100%. Asample represents the absorbance of the experimental well (containing cells, CCK-8 solution, medium, and peptide diluent); Abaseline represents the absorbance of the baseline well (containing no cells, CCK-8 solution, medium, and peptide diluent); Acontrol represents the absorbance of the control well (containing cells, CCK-8 solution, medium, and no peptide diluent); and Ablank represents the absorbance of the blank well (containing no cells, CCK-8 solution, medium, and no peptide diluent).
7. Coculture of Jurkat T cells with Cal27 cells for analysis of Jurkat T cell activity
Jurkat T cells were stimulated with PMA (1652981, 50 ng/ mL, PeproTech) and ionomycin calcium salt (ionomycin, 5608212, 1 μg/mL, PeproTech) for 24 hours. Cal27 cells were stimulated with 500 U/mL IFN-γ (300-02, PeproTech) for 48 hours. Stimulated Jurkat T cells were pre-incubated with nABP284, nABPD1 (10 μM) or a functional-grade PD-1 monoclonal antibody (16-9989-82, 2 μg/mL, J116, eBioscience, Thermo Fisher Scientific, Waltham, MA) for 1 hour at 37°C. Subsequently, 1×104 Cal27 cells were seeded in 96-well plates, and after the Cal27 cells had adhered, the supernatant was discarded, and Jurkat T cells were added to the wells at a ratio of 4:1 with Cal27 cells in 200 μL of medium. The supernatant was collected after 24 hours of coculture, and the IL-2 levels were measured with an IL-2 Human Uncoated ELISA (enzyme-linked immunosorbent assay) kit (88-7025-86, Thermo Fisher Scientific) according to the manufacturer’s instructions. Finally, the absorbance was determined with a Victor X5 Multilabel Plate Reader (PerkinElmer) at 450 nM.
8. Coculture of ICIK cells with Cal27 cells for analysis of ICIK cell-mediated lethality
After mixing human peripheral blood with PBS at a ratio of 1:1, the mixture was added on top of the same volume of Ficoll solution (17-1140-02, GE Healthcare). Density gradient centrifugation (400 ×g, 20 minutes) was performed to separate the components of the human peripheral blood. After isolation, PBMCs were added to Petri dishes coated with 10 μg/mL GMP-grade anti-CD3 mAb (clone OKT3, T210, Takara, Tokyo, Japan) and incubated at 4ºC overnight; then, the cells were cultured in RPMI 1640 (Sigma-Aldrich) supplemented with 10% fetal bovine serum (10099-141, Gibco), IFN-γ (300-02, 2000 U/mL, PeproTech), and IL-2 (200-02, 10 ng/mL, PeproTech). The medium was changed every 3 days, and the cells were cultured for 10 days. We called these activated PBMCs ICIK cells. To analyze ICIK cell-mediated killing, Cal27 cells were stimulated with 500 U/mL IFN-γ (300-02, PeproTech) for 48 hours, and ICIK cells were pre-incubated with nABP284, nABPD1 (10 μM), or a functional-grade PD-1 monoclonal antibody (16-9989-82, 2 μg/mL, J116, eBioscience, Thermo Fisher Scientific) for 1 hour at 37°C. Subsequently, 1×104 Cal27 cells were seeded in 96-well plates. After the Cal27 cells had adhered, the supernatant was discarded, and ICIK cells were added to the Cal27 cells in each well at a ratio of 10:1 in 100 μL of medium. The 96-well plates were centrifuged at 250 ×g for 4 minutes to ensure that the ICIK cells and Cal27 cells came into contact with each other. After 6 hours of incubation, lactate dehydrogenase (LDH) levels in the medium were measured with a CytoTox 96 Non-Radioactive Cytotoxicity Assay Kit (G1780, Promega, Madison, WI) according to the manufacturer’s instructions, and the absorbance was measured with a Victor X5 Multilabel Plate Reader (PerkinElmer) at 490 nM.
Results
1. Optimization of nABP284 and design of nABPD1
(1) Design of nABPD1
The amino acid sequence of nABP284 was determined by phage display technology in our previous experiments. By analyzing the structure of the complex of PD-1 and its ligand PD-L1 in RCSB PDB (4ZQK) and comparing the sequence homology of human PD-L1 and nABP284, we found that the 2RLKEIA7 motif of nABP284 is homologous to the 125RKYDA121 sequence in the extracellular domain of human PD-L1, which is one of the key regions at which PD-1 and PD-L1 interact, as reported in the literature. In particular, the A121, K124, and R125 residues of this sequence are crucial determinants of PD-1 and PD-L1 binding (Fig. 1A). Furthermore, on the basis of the cleft between Y68 and E136 near this key region in PD-1, we designed an “SHHHRL” motif, whose imidazole may have the potential to combine with this hot spot. Computer-simulated structural analysis using Autodock provided evidence for this hypothesis (Fig. 1B). When binding to PD-1, the side chain of nABPD1 (SHHHRL) with HHH can complement the main chain to enhance affinity. In conclusion, we obtained a new peptide, nABPD1, by comparing the sequence homology of nABP284 and PD-L1 and adding “HHH”-containing amino acid chains (SHHHRL) to the region that may bind to PD-1 (Fig. 1C).
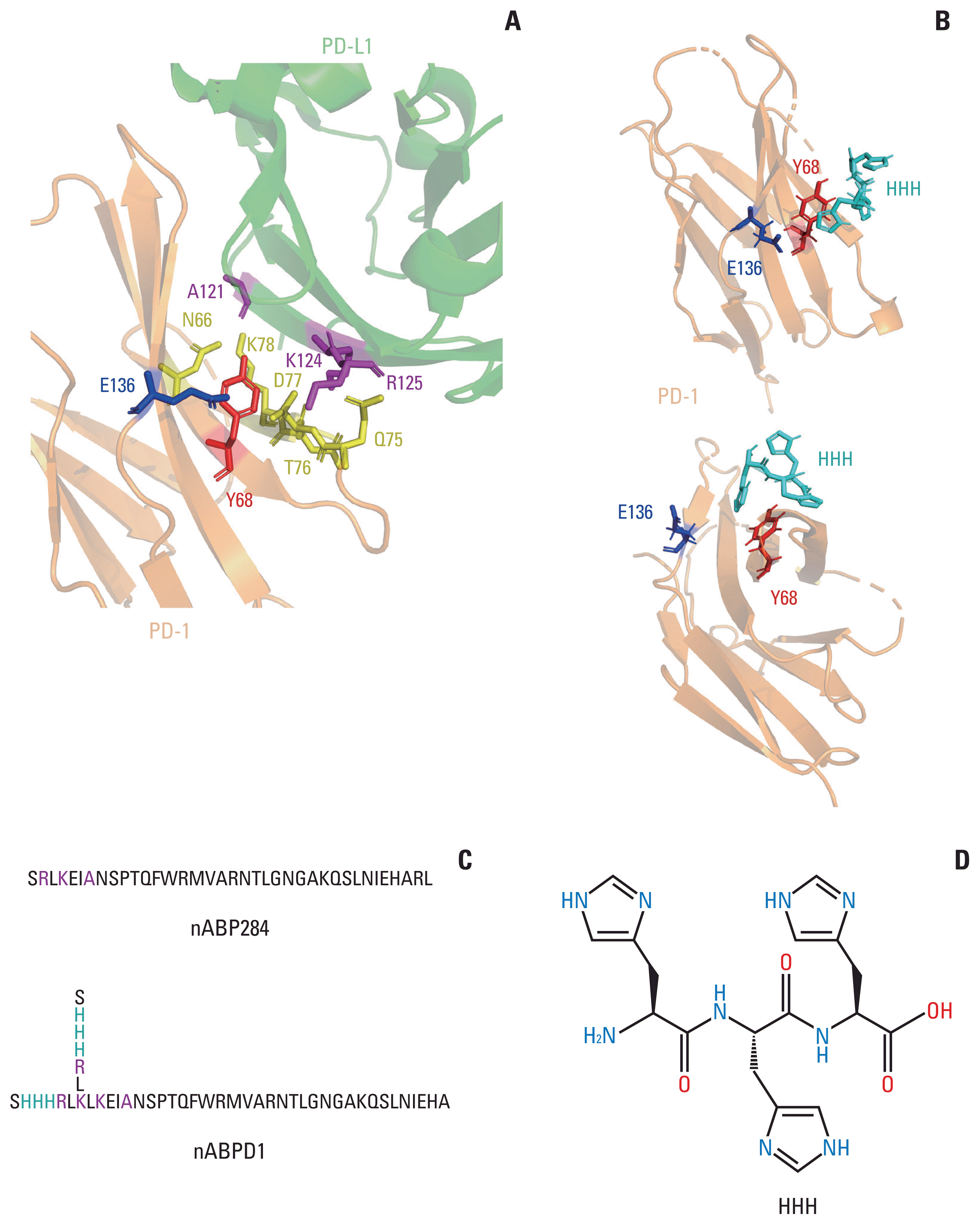
Fig. 1
Optimization and improvement of the peptide. (A) Main binding regions between human programmed cell death protein 1 (hPD-1) and human programmed death-ligand 1 (hPD-L1). The key amino acid residues through which it binds programmed cell death protein 1 (PD-1) are labeled. The region including A121 K124 R125 in PD-L1 shows sequence homology with nABP284. (B) AutoDock simulation provided evidence for the hypothesis that the imidazole ring on histidine could bind to the cleft between Y68 and E136 in PD-1. (C) Sequence of nABP284 and nABPD1, nABPD1 was optimized from nABP284 by adding branched chains (SHHHRL) with three consecutive histidine residues (HHH) on the N-termini. (D) Structural formula of HHH.
![]()
(2) Synthesis of peptides
2. Binding specificity and affinity of the new peptide
We determined that the KD value of nABPD1 was 11.9 nM (Fig. 2B), which was significantly higher than that of nABP284 (Fig. 2A). Unstimulated Jurkat T cells expressed very low levels of PD-1; conversely, PMA-stimulated Jurkat T cells showed high PD-1 expression (Fig. 3A). To demonstrate the binding specificity of nABPD1 to PD-1, we allowed nABPD1 to bind to Jurkat T cells before and after stimulation. As exhibited in Fig. 3A, nABPD1 could bind to Jurkat T cells in which PD-1 expression was stimulated by PMA and ionomycin, while very little nABPD1 bound to unstimulated Jurkat T cells. The new peptide had a higher rate of binding to PD-1–expressing Jurkat T cells than nABP284, and this increase in the binding rate was dose-dependent (Fig. 3B and C). These results indicate that the binding of nABPD1 to PD-1–expressing Jurkat T cells is specifically mediated by PD-1.
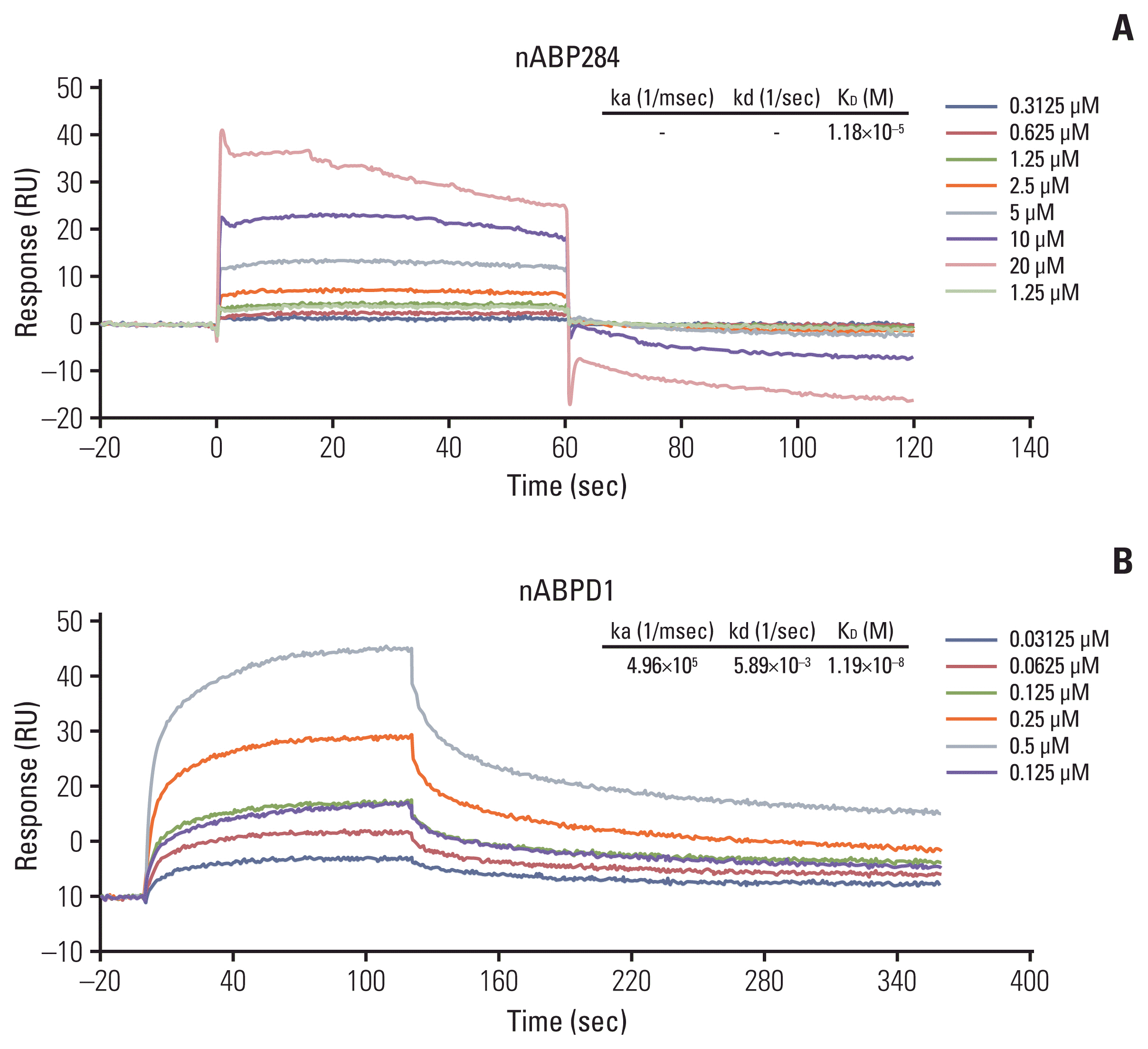
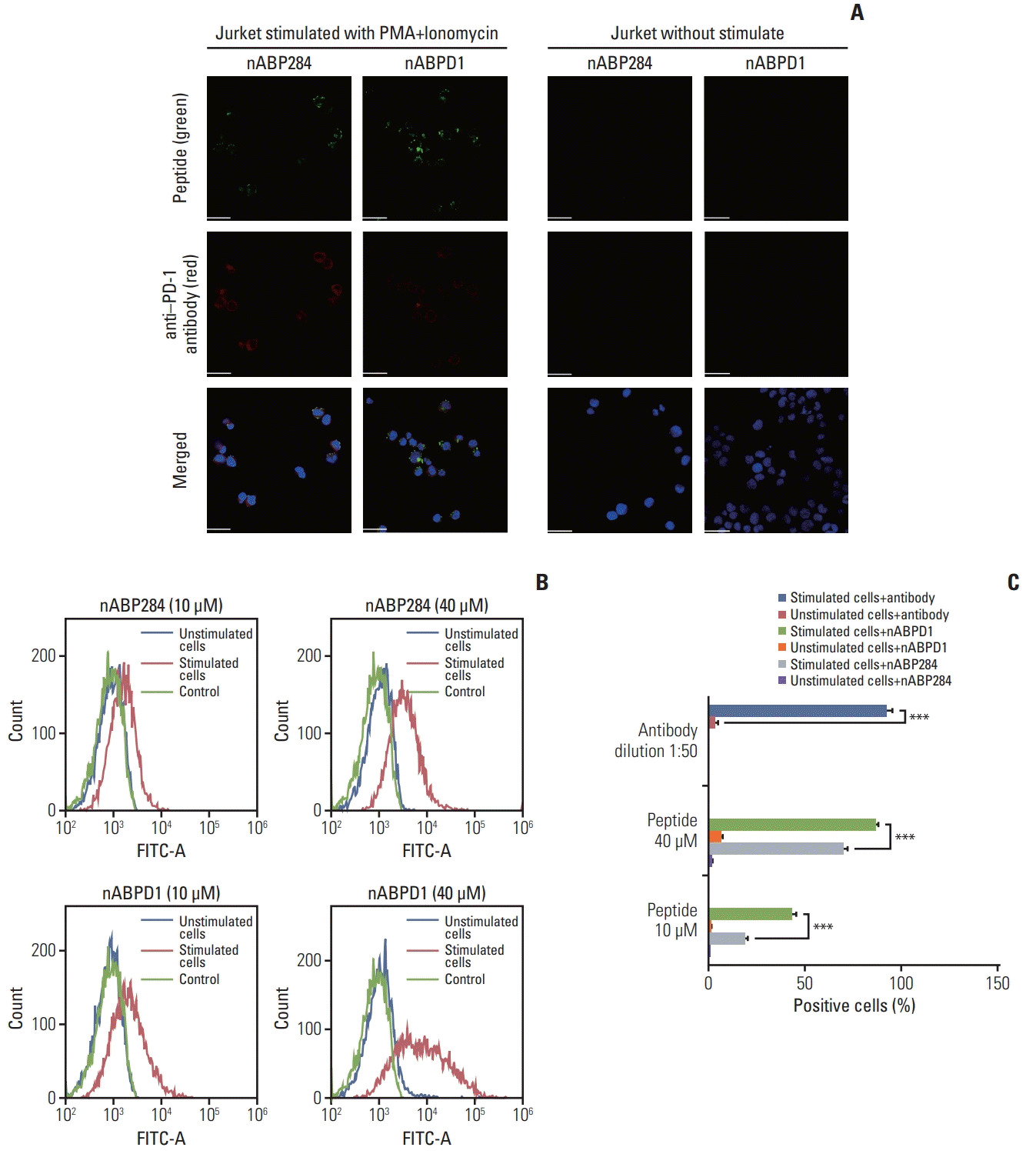
Fig. 2
Binding affinity of the peptides. The binding affinity of nABP284 (A) and nABPD1 (B) to human programmed cell death protein 1 (PD-1) recombinant protein was analyzed by surface plasmon resonance, and the affinity of nABPD1 for PD-1 was significantly enhanced compared with that of nABP284.
![]()
Fig. 3
Binding of the peptides to stimulated Jurkat T cells. (A) Expression of programmed cell death protein 1 (PD-1) and binding of 10 μM nABP284 or nABPD1 labeled with FITC (green) on Jurkat T cells before and after treatment with PMA (phorbol 12-myristate 13-acetate; 50 ng/mL) and ionomycin (1 μg/mL) for 24 hours. After allowing the peptide to bind, the cells were incubated with an anti–PD-1 antibody (red), and nuclei were stained with DAPI (blue). Images were merged. (B, C) Flow cytometry analysis of the binding of nABP284 or nABPD1 labeled with FITC to Jurkat T cells before and after stimulation. The data are presented as the mean±standard error of three independent experiments and were analyzed by Student’s t test. ***p < 0.001.
![]()
3. Blocking and neutralizing capacity of the new peptide
The purpose of these studies was to determine whether the enhanced binding ability of the new peptide compared with that of nABP284 resulted in enhanced blocking and neutralizing capacity. Jurkat T cells were pre-stimulated with PMA and ionomycin for 24 hours. After 1 hour of incubation with nABP284, nABPD1, or a functional anti–PD-1 antibody, the amount of PD-1 that was bound on the surface was reduced in treated Jurkat T cells compared with untreated Jurkat T cells (Fig. 4A). Quantification of the mean fluorescence intensity indicated that both nABP284 and nABPD1 had the ability to block PD-1 on the surface of stimulated Jurkat T cells and that nABPD1 had a better blocking capacity than nABP284 at the same concentration (Fig. 4B).
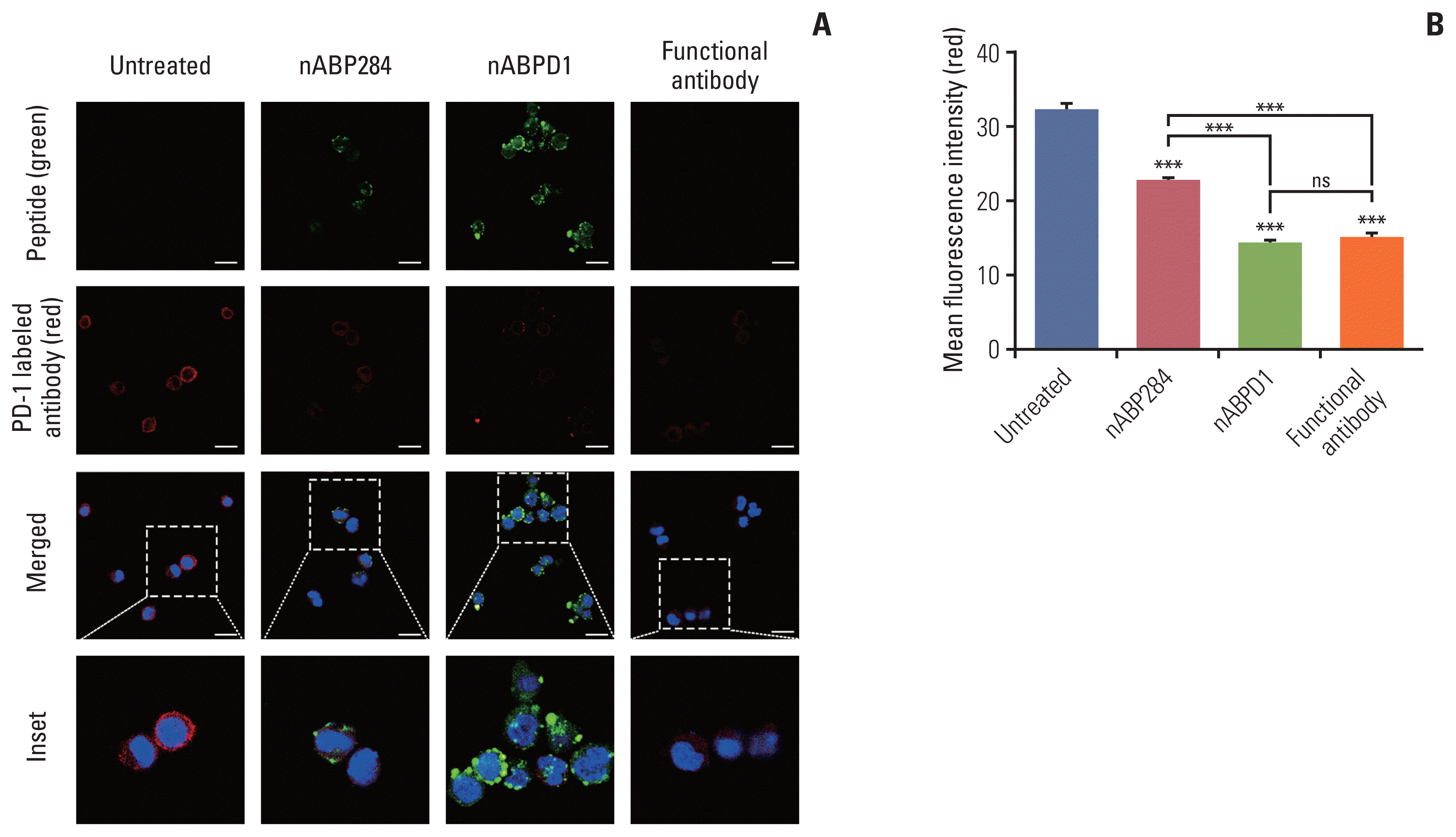
Fig. 4
Competition of peptides for binding to programmed cell death protein 1 (PD-1). (A) Images of the binding of nABP284, nABPD1 and an anti–PD-1 antibody to PD-1 expressed on the surface of Jurkat cells stimulated with PMA (phorbol 12-myristate 13-acetate) and ionomycin for 24 hours were obtained by confocal microscopy. Scale bars=12 μm. (B) Image J was used to measure the mean fluorescence intensity to evaluate PD-1 binding on the surface of stimulated Jurkat cells that were blocked with nABP284, nABPD1, or a functional PD-1 antibody in advance. The data are presented as the mean±standard error of three independent experiments and were analyzed by one-way ANOVA. ***p < 0.001; ns, not significantly different.
![]()
Next, we examined the ability of nABP284 and nABPD1 to neutralize recombinant human PD-L1 protein. As the ligand for PD-1, PD-L1 can specifically bind to PD-1. Before allowing it to bind stimulated Jurkat T cells expressing PD-1, we mixed nABP284 or nABPD1 with recombinant human PD-L1 protein at various concentrations. The binding rate of the recombinant human PD-L1 protein was determined by flow cytometry, and at concentrations of 4, 13.3, and 40 μM, the neutralization capacities of nABPD1 and nABP284 were significantly different (Fig. 5A); additionally, the binding of human PD-L1 recombinant protein to stimulated Jurkat T cells overexpressing PD-1 was inhibited by increasing the concentration of nABP284 or nABPD1. The half maximal inhibitory concentrations (IC50) of nABP284 and nABPD1 were 9.610 and 2.585 μM, respectively (Fig. 5B). The neutralization capacity of nABPD1 was significantly enhanced compared with that of nABP284 (Fig. 5A and B).
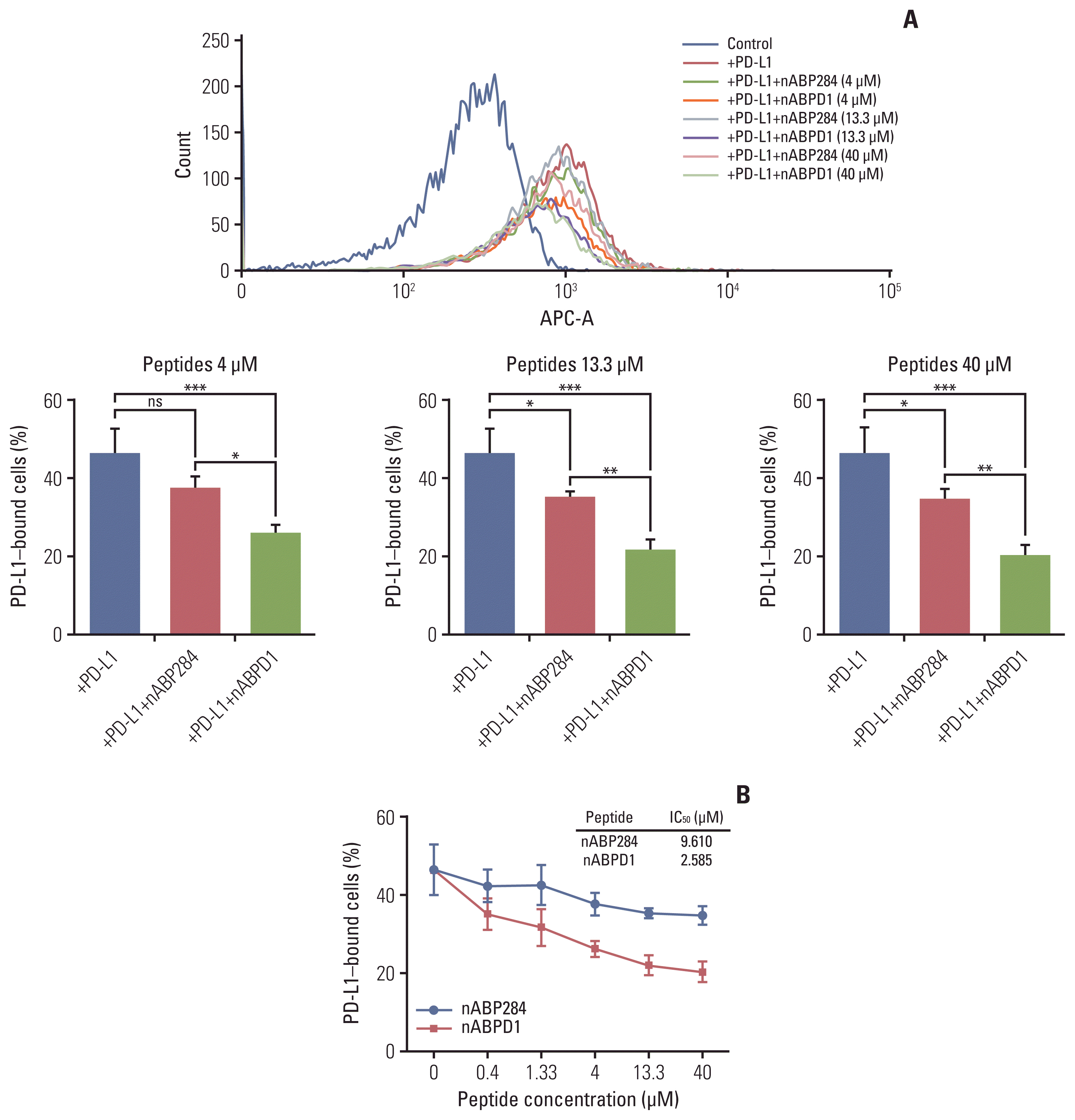
Fig. 5
Neutralization of programmed death-ligand 1 (PD-L1) with the peptides. (A) The neutralization capacities of nABP284 and nABPD1 at concentrations of 4, 13.3, or 40 μM. (B) Flow cytometry showed that the binding of human PD-L1 recombinant protein to stimulated Jurkat cells overexpressing programmed cell death protein 1 was inhibited by increasing the concentrations of nABP284 or nABPD1. nABPD1 showed a stronger blocking effect than nABP284 at the same concentration. The IC50 values of nABP284 and nABPD1 were 9.610 and 2.585 μM, respectively. The concentration of PD-L1 was 2 μg/mL. The data are presented as the mean±standard error of three independent experiments and were analyzed by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significantly different.
![]()
4. The new peptide rescues the inhibitory effect of Cal27 cells on Jurkat T cells
After stimulation of Jurkat T cells with PMA and ionomycin, PD-1 was highly expressed on the surface of the cells (Fig. 3A). Furthermore, Jurkat T cells secreted a large amount of IL-2. Similarly, the expression of PD-L1 on the surface of Cal27 cells increased after pre-incubation with increasing IFN-γ concentrations (Fig. 6A). The cytokine secretion level is an important indicator of T cell function. After coculture, Cal27 cells inhibited Jurkat T cells through the interaction of PD-1 with PD-L1, which was manifested by a decrease in IL-2 levels in the medium, as determined by ELISA (Fig. 6B). To examine whether the new peptide is more effective than nABP284 in blocking the interaction between PD-1 and PD-L1, we first determined that nABP284 and nABPD1 were not toxic to Cal27 cells and Jurkat T cells (Fig. 6C), and then, we added nABP284, nABPD1, or a functional anti–PD-1 antibody to the coculture system. The results showed that the IL-2 level was significantly increased in the coculture systems to which nABP284, nABPD1, or the anti–PD-1 anti-body was added compared to the control coculture system (Fig. 6B). Hence, nABP284 and the new peptide rescued the inhibitory effect on Jurkat T cells by blocking the interaction of PD-1 and PD-L1, and the blocking effect of nABPD1 was significantly better than that of nABP284.
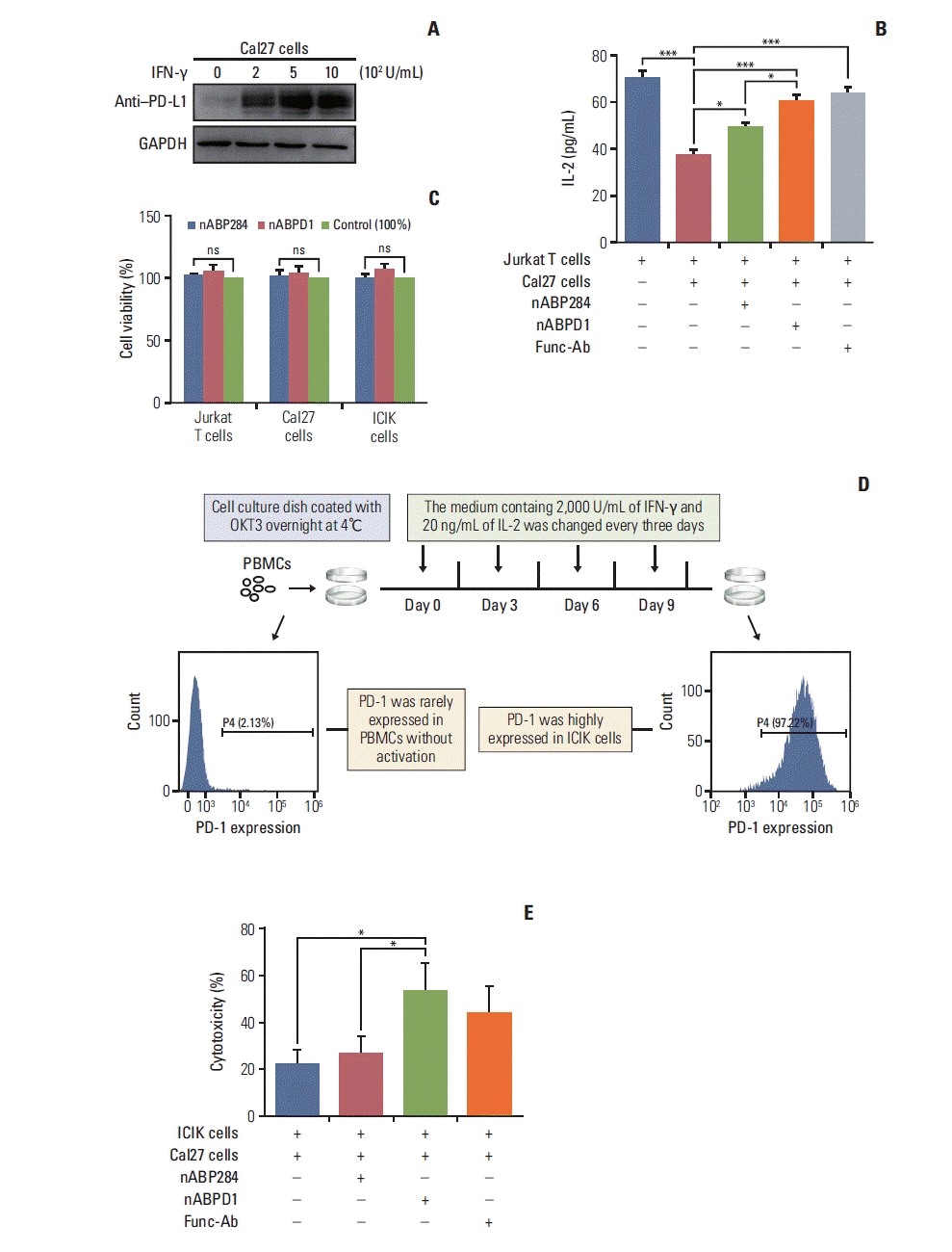
Fig. 6
In vitro analysis of peptide function. (A) Expression of programmed death-ligand 1 (PD-L1) on cal27 cells after 48 hours of stimulation with different concentrations of interferon γ (IFN-γ). (B) Coculture with Cal27 cells significantly reduced the amount of interleukin 2 (IL-2) secreted by Jurkat T cells, and this inhibitory effect was reversed by nABP284 or nABPD1. nABPD1 showed a better effect than nABP284. (C) The Cell Counting Assay (Cell Counting Kit-8) revealed that nABP284 and nABPD1 were not directly cytotoxic to stimulated Cal27 cells, Jurkat T cells, or improving cytokine-induced killer (ICIK) cells at a concentration of 64 μM after 24 hours of culture. (D) Venous blood was drawn from donators, and peripheral blood monocytes (PBMCs) were isolated from the venous blood by density gradient centrifugation with Ficoll. OKT3, IL-2, and IFN-γ were used as shown in the image above to stimulate PBMCs. These stimulated PBMCs were called ICIK cells. The expression of programmed death 1 (PD-1) on PBMCs and ICIK cells was analyzed by flow cytometry. (E) Compared with untreated group and nABP284 group, nABPD1 significantly enhanced the cytotoxicity of ICIK cells to Cal27 cells. The data are presented as the mean±standard error of three independent experiments and were analyzed by one-way ANOVA. *p < 0.05, ***p < 0.001; ns, not significantly different.
![]()
5. The new peptide enhances the cytotoxicity of ICIK cells to cancer cells
As shown in Fig. 6D, PBMCs were isolated from peripheral blood drawn from volunteers and activated by stimulation with a series of cytokines, including OKT-3, IL-2, and IFN-γ. We called these activated PBMCs ICIK cells. Flow cytometry showed that ICIK cells significantly overexpressed PD-1 compared with freshly isolated PBMCs (Fig. 6D). ICIK cells were cocultured with IFN-γ–stimulated Cal27 cells, and the cell-mediated cytotoxicity of ICIK cells to Cal27 cells was evaluated by measuring the concentration of LDH in the medium by ELISA. Due to the PD-1/PD-L1 pathway, the cell-mediated cytotoxicity of ICIK cells to Cal27 cells was relatively weak (Fig. 6E). The cytotoxicity of ICIK cells to Cal27 cells was significantly increased in the nABPD1-treated group compared with the control group and nABP284 group (Fig. 6E). Considering that nABPD1 has no direct ability to kill Cal27 cells (Fig. 6C), the above experimental results suggest that nABPD1 can enhance ICIK cell-mediated cytotoxicity by blocking the interaction between PD-1 and PD-L1 (Fig. 6D and E).
Discussion
For immune checkpoint inhibitors, affinity for the target is crucial. Having a better affinity for immune checkpoint ligands gives immune checkpoint inhibitors an advantage over the proteins with which they compete [21]. Due to the limitations of phage display technology, such as the lack of diversity in peptide libraries [22], peptides identified by phage display technology usually must be further optimized. Unlike the complex structures of monoclonal antibodies, the simple structures of small molecules such as peptides allow directed evolution through computer simulations. Instead of expanding the diversity of the peptide library and repeating biopanning, it is more efficient to improve peptides with less than ideal affinity through this type of optimization method.
In our study, after sequence alignment and computer simulations, we predicted the possible sequence through which nABP284 binds to PD-1 and designed side chains (SHHHRL) with three consecutive histidine residues (HHH) to increase the affinity of the peptide for PD-1 based on potential hot spots near the binding region. The structure of three consecutive histidine residues (HHH) has also been reported to be the key structure that enables V-domain immunoglobulin suppressor of T cell activation to bind to target molecules [23]. The optimized peptide (nABPD1) possesses better affinity for PD-1 (11.9 nM) than nABP284; in contrast, in terms of affinity, nABP284 (11.8 μM) has no significant advantage over the PD-L1 protein (8.2 μM) [24]. The above series of experiments proved that nABPD1 is significantly more efficient than nABP284 in competing with the PD-L1 protein to bind to PD-1 and in blocking the PD-I/PD-L1 interaction. These results indicate that our method for optimizing peptides identified by phage display technology is feasible.
Our previous studies have shown that ICIK cell therapy could restore immune function and prolong survival in patients with head and neck squamous cell carcinoma [19,20]. ICIK cells were transformed from human PBMCs by cytokine-induced transformation. PD-1 expression on the surface of human PBMCs was significantly increased during cytokine-induced transformation to ICIK cells. In the above in vitro experiments, nABPD1 significantly enhanced the lethality of ICIK cells to Cal27 cells. Thus, nABPD1 could be cultured with ICIK cells in vitro before ICIK cell treatment. By blocking PD-1 on the surface of ICIK cells, nABPD1 reversed the inhibitory effect of the interaction between PD-1 and PD-L1.
In vivo stability and half-life are important criteria for peptide immune checkpoint inhibitors [25]. An increase in molecular weight generally increases the stability of a peptide in vivo [26]. Unlike nABP284, nABPD1 has a branched chain (SHHHRL), making the secondary structure of the new peptide more complex. We hypothesized that the stability of the new peptide would also benefit from this modification [27,28]. Due to their vigorous metabolism, the local microenvironment of solid malignant tumors is mostly acidic, and as a basic peptide, nABPD1 tends to accumulate in the acidic microenvironment of tumors [29]. Furthermore, nABPD1 has five positive charges under physiological conditions and can easily bind to negatively charged heparin D sulfate on the tumor cell membrane through electrostatic interactions. The peptide’s capacity for deep tissue penetration makes this phenomenon more likely to occur. Therefore, we hypothesize that the new peptide (nABPD1) possesses the ability to target tumor tissue. All these assumptions need to be confirmed by further experiments.
The use of multifunctional antibodies has been proven to be feasible in recent experiments [30]. Such multitargeting molecules enhance contact between target cells while acting as their own immune checkpoint inhibitors. Peptides, as small molecules that are easy to modify and inexpensive to synthesize, are potential candidates for the multifunctional molecules mentioned above. Considering that nABPD1 possesses a high affinity for PD-1, peptides with high affinity for PD-L1 could be combined with nABPD1, and a multifunctional peptide targeting both PD-1 and PD-L1 might induce the aggregation of T lymphocytes in tumor tissues while blocking the interactions between PD-1 and PD-L1. We will focus on this in our future research.
Taken together, our results identified a promising PD-1-blocking peptide (nABPD1), and our approach to modifying nABP284 also provided new ideas for optimizing target-binding peptides identified by phage display technology. These results lay the foundation for our future research.
 XML Download
XML Download