

 PDF
PDF Citation
Citation Print
Print



Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative condition characterized by loss of motor neurons in the cerebral motor cortex, brainstem, and spinal cord. It is typically heralded by motor weakness in the limbs and/or bulbar muscles and results in progressive muscle weakness beyond the site of initial symptoms, resulting in a fatal outcome generally within 2 to 4 years from the onset of disease [1]. Diagnosis of the disease is based on clinical and electrophysiological evidence of upper motor neuron (UMN) and lower motor neuron (LMN) involvement and the distribution of clinical signs of the disease (e.g., craniobulbar, cervical, thoracic, and lumbosacral regions) [2-4]. Notably, ALS should be considered a syndrome with a broad spectrum rather than a single disease entity because its symptoms and clinical course are very diverse (e.g., UMN vs. LMN predominance, bulbar vs. limb [spinal] predominance, rapid vs. slow progression, and the extent of concomitant cognitive impairment) [5]. ALS was once considered a pure motor disorder with ‘3 Ps’ (progressive, painless, and paralysis); however, a growing body of evidence indicates that ALS is a multisystem disease with early and diverse non-motor symptoms including cognitive and behavioral changes, neuropsychiatric disturbances, sleep disruption, excess secretions, metabolic abnormalities, bowel and bladder dysfunction, changes in bone health, olfactory and somatosensory impairment, and pain [6]. These non-motor symptoms of ALS have attracted research attention over the last decade, with the identification of a repeat expansion of the C9orf72 gene in some patients and many shared pathologies across the spectrum of ALS and frontotemporal dementia in these patients [7,8].
The active study of pain in ALS, as well as other non-motor symptoms, began approximately 10 years ago, although there were earlier observations [9-11]. Previous studies revealed that these symptoms are prevalent among patients with ALS and negatively impact the lives of these patients and their caregivers, physically, psychologically, socially, and spiritually [12-15]. In addition, the two major guidelines on the management of ALS (from the American Academy of Neurology [AAN] and European Federation of Neurological Societies [EFNS]) include the treatment of pain in these patients [16,17], and there has been a Cochrane review on this subject [18]. However, a more thorough understanding of the prevalence, clinical characteristics, underlying mechanisms, and effective management of pain in this population is required to improve clinical practice and patient outcomes. In this review, the epidemiology of pain in patients with ALS, its clinical characteristics, and suggested underlying mechanisms of pain will be discussed before summarizing the assessment and treatment strategies for pain in ALS.
Epidemiology of pain in amyotrophic lateral sclerosis
The prevalence of pain in patients with ALS varies greatly from as low as 15% to as high as 85% [10,13,15,19-24]. These differences are thought to arise from the different study designs and settings, definitions of pain, and tools used to assess pain. A recent meta-analysis on the prevalence of pain in ALS estimated the pooled prevalence to be 60% (95% confidence interval [CI], 50%–69%) with a high degree of heterogeneity (I2=94%, p<0.001) [25]. This meta-analysis also found that studies using tailored measures (e.g., clinical interviews, unvalidated questionnaires, or retrospective reviews of the medical record) to assess pain tended to show a lower pooled prevalence of 47% (95% CI, 34%–60%) and greater heterogeneity (I2=90%, p<0.001) compared to those using tailored measures (I2=82%, p=0.002, respectively), while studies using only validated measures showed a pooled pain prevalence of 65% (95% CI, 56%–73%, similar to the overall pooled prevalence of 60%) and lower heterogeneity. This demonstrates the importance of appropriate assessment tools for pain. Nevertheless, the two major guidelines for the management of ALS do not suggest which assessment tools should be selected. Another study showed that less than 20% of ALS clinics used questionnaires involving pain rating scales other than just open-ended questions [19]. Therefore, the establishment of a standardized assessment tool for pain in ALS is necessary to better manage pain in patients with ALS.
Currently, the most frequently used formal assessment tool is the Brief Pain Inventory questionnaire [13,15,19,22,24], which assesses current pain, its intensity over the previous week, and the extent of interference with mood, sleep, and physical, working, and social activities. Other validated assessment tools include the Neuropathic Pain Scale, short-form McGill Pain Questionnaire, Neuropathic Pain Symptom Inventory, and Neuropathic Pain Diagnostic Questionnaire [20,23]. However, although these tools are validated and are useful in discriminating and describing pain, it is unclear how useful they are for assessing pain in patients with ALS, who often have more than one type of pain [26]. Therefore, the development of standardized assessment tools for the diagnosis and monitoring of pain in ALS is important for improving patient care and outcomes.
In addition, the attention that physicians and patients pay to pain might also result in an inconsistency in its reported prevalence [26]. For example, a recent study conducted by Åkerblom et al. [12] indicated that some patients with ALS who were experiencing pain did not report it to their physician because they thought pain was not as important as other symptoms of the disease, they had received insufficient attention from the physician, or they believed there was no effective pain treatment, just as there was no effective disease treatment. Considering these points, it is important not only to evaluate pain at every visit using a structured assessment tool but also to train healthcare professionals accordingly.
Characteristics of pain
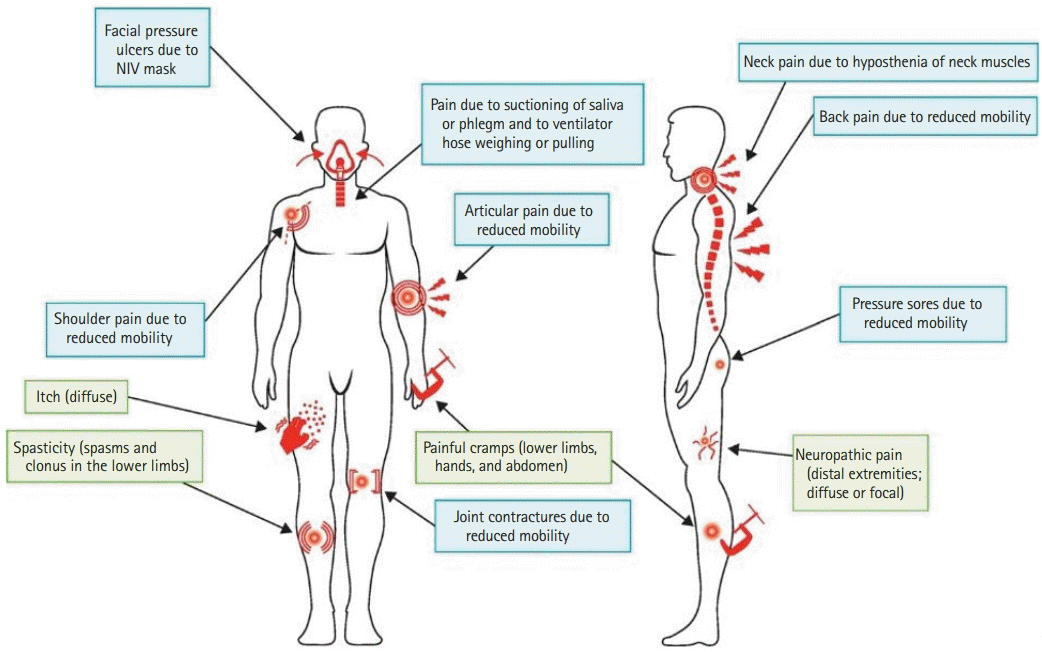
Primary pain in patients with ALS includes neuropathic pain and pain from spasticity or cramps. Secondary pain is mainly nociceptive, occurring with the progression of muscle weakness and atrophy, prolonged immobility causing degenerative changes in joints and connective tissue, and long-term home mechanical ventilation (HMV) either noninvasively by mask interface or invasively by tracheostomy (Fig. 1) [26,27].
Pain in ALS is largely nociceptive in nature, although it cannot explain all types of ALS pain. Neuropathic pain has not been reported to be prevalent among patients with ALS [16,26,28-30]. Previous studies have indicated a prevalence of neuropathic pain of 9% [28], 1% [29], and 5% [30] using Douleur Neuropathique 4 (DN4) as a screening tool; these values are not higher than those of the general population, ranging from 6.9% to 10.0%, as reported in a previous systematic review [31]. However, a recent study indicated that 62.5% of patients with ALS experienced neuropathic pain, described as electric shock, burning, dull, stabbing, throbbing, painful cold, sharp, or shooting [32]. However, that study did not adopt a validated measure for neuropathic pain. Although neuropathic pain seems to be rare in ALS, this finding might result from the fact that DN4 is only a screening tool for neuropathic pain and may not be a definite measure to diagnose neuropathic pain in ALS [28]. Furthermore, the low prevalence of neuropathic pain might have occurred because a large number of patients were already taking neurotropic medications such as riluzole, the only drug approved by the U.S. Food and Drug Administration for ALS treatment, which blocks the presynaptic release of glutamate [33] and thereby might reduce neuropathic pain [29,34,35]. Some neuropathic pain might be due to spinal disorders, at least partially, considering that the peak incidence of ALS occurs between 58 and 63 years of age [1], an age group in which spinal disorders are common. Although it has been reported that 14% to 32% of the diagnostic delay in ALS is due to the misdiagnosis of ALS as a spinal disorder such as myelopathy or radiculopathy [36-42], no study has evaluated the exact prevalence of spinal disorders in patients with ALS.
Other sources of primary pain include cramps and spasticity. Muscle cramp refers to a sudden and involuntary muscle contraction originating from the peripheral nerves [43] and is reported to be a major source of pain in patients with ALS, affecting approximately two-thirds of patients [11,44]. In a previous study, which followed 41 patients with ALS for up to 21 months, 95% of the patients had experienced cramps over the course of the disease [45]. Muscle cramps in patients with ALS are believed to originate from instability of the affected motor unit and are typically associated with denervation of the muscles [43]; patients with limb-onset ALS are more frequently affected by them [43,45]. Spasticity is a velocity-dependent increase in muscle tone, which results from the loss of inhibitory control of UMNs, causing stiffness and spasms in the affected muscles as well as difficulties in fine motor control [46]. According to a recent study conducted in a tertiary hospital in France, the prevalence of spasticity was 36% in patients with ALS, and approximately half (42.5%) of those patients had mild pain. However, 16.7% of patients with spasticity had moderate to severe pain, with numeric rating scale scores of ≥4 [47].
Secondary pain is mainly nociceptive, which means it arises from nonneuronal tissue damage or the activation of peripheral nociceptors as a result of mechanical or other noxious stimuli [48]. Secondary pain is known to develop as ALS progresses. Joint pain, which is prevalent in patients with ALS, develops when weakened and wasting muscles are unable to provide support to the joint [26,49]. It has been reported that shoulders and hips are the most frequently affected joints [13,50]. Immobility might cause skin pressure, which might be perceived as pain, although pressure sores do not occur frequently despite the poor mobility of patients with ALS [51]. Long-term HMV may be another source of pain. In patients undergoing noninvasive ventilation (NIV), skin problems due to mask interfaces, especially on the nasal bridge, can cause pain [26]. In invasively ventilated patients with a tracheostomy who remain in the same position over a period of time during HMV, irritation of the throat from the weight of the circuits (tubes) and suctioning of the secretions can result in significant discomfort and pain, which are often unnoticed by caregivers or medical professionals [52].
In terms of the localization of pain, previous studies have not found a specific pattern, and pain can involve any part of the body, including the proximal and distal sites of the upper and lower extremities and the back, or it could be diffuse [13,15,19,22,23,53]. A recent meta-analysis, which analyzed a total of 393 patients with ALS who reported 715 locations of pain, reported that the most commonly reported location was the upper limbs (including shoulders and extremities) (41.5%), followed by the lower limbs (33.7%), and the head, neck, trunk, and back (24.8%) [25].
The intensity of pain was also reported in the same study; among 1,426 patients who reported pain intensity, 78.8% reported moderate pain, 17.5% reported severe pain, and only 2.0% and 1.7% reported mild and very severe pain, respectively [25], which is consistent with previous reports [23,29]. However, other studies have indicated that the intensity was mainly mild [14,22]. Again, this inconsistency might result from the different study designs and settings, the definition of pain, and the assessment tools for pain, as well as the cross-sectional nature of the aforementioned studies. A recent longitudinal study indicated that at least moderate average pain was reported in about two-thirds of patients who completed a 1-year follow-up [54], indicating that moderate to severe pain might be persistent in patients with ALS.
Several studies have indicated the occurrence of pain early in the disease course [9,15,19,26,32,55]. In a study including 424 patients, 34% of patients with ALS reported pain in the early stage of disease [19], and a recent epidemiological study revealed that patients with ALS had been prescribed more drugs for neuropathic pain (hazard ratio, 1.84; 95% CI, 0.99–3.42) up to 2 years before the onset of ALS [56]. The question of whether pain worsens with ALS progression varies widely among studies [13-15,22,23,53], probably because of the cross-sectional nature of these studies [26]. A longitudinal study indicated that pain intensity increased by 1 point on the visual analog scale from the first visit to the last, with a median follow-up period of 104 days (range, 35–846 days) [57]. Similarly, a recent study including 151 patients at baseline also revealed that the intensity and quality of pain and the impairment it causes did not change significantly over time [54], which is consistent with findings from another longitudinal study [58].
Treatment of pain in amyotrophic lateral sclerosis
Currently, there is little high-quality evidence regarding the pharmacological treatment of pain in ALS, as there are no randomized or quasi-randomized treatment trials of pain in ALS. However, several guidelines on the clinical management of ALS include management of pain, cramps, and spasticity [16,17,59], and there are Cochrane reviews [18,60]. Prior to treatment, the exact patterns and causes of pain must first be identified; then, the treatment should be tailored to each patient. Treatment options can be classified into pharmacological and nonpharmacological treatments, for which pharmacological treatment is known to be more effective for the primary pain, whereas nonpharmacological approaches are regarded as more effective for secondary pain in ALS [26]. A summary of interventions for the management of pain according to its etiology is presented in Table 1, including both self-treatment and professional treatment.
1. Treatment of primary pain in amyotrophic lateral sclerosis
As stated earlier, neuropathic pain does not appear to be prevalent in patients with ALS; however, this type of pain is significant because it can be controlled according to pharmacological management pathways for neuropathic pain [61]. The most widely used drugs for the treatment of neuropathic pain are gabapentin, pregabalin, and tricyclic antidepressants [49]. Although opioids are not recommended as first-line therapy, they can be used when pain is not controlled or in advanced stages in cases of increased pain or with symptoms related to respiratory insufficiency, such as dyspnea and sleep disturbance [62]. However, there is little evidence to support the safety and efficacy of opioid use [18].
Quinine sulfate is the most frequently prescribed drug in European countries, although its use for cramps is proscribed by the U.S. Food and Drug Administration [26,63]. It should also be noted that quinine sulfate can cause serious side effects, such as thrombocytopenia and drug interactions [49]. Mexiletine at a dose of 300 mg per day and dronabinol at a dose of 5 mg twice daily could be used as first-line therapies; gabapentin and levetiracetam are suggested as second-line therapies in the guidelines [16,17].
Spasticity can be effectively managed with appropriate medication. Although there are no controlled clinical trials in patients with ALS, baclofen, tizanidine, benzodiazepines, dantrolene, and carbamazepine can be used to reduce spasticity [49]. However, undesirable side effects, such as weakness, daytime somnolence, or excessive fatigue, are common with these medications. Therefore, careful dose titration and appropriate physiotherapy are required [50,64,65]. In cases of refractory spasticity, intrathecal baclofen pump placement was proposed as an option, and a previous study indicated an average reduction of 54% in pain intensity after the procedure. However, these findings should be interpreted with caution considering the open-label study design and lack of follow-up evaluation [66].
2. Treatment of secondary pain in amyotrophic lateral sclerosis
Musculoskeletal pain secondary to progressive wasting and weakness of the muscles should be managed with careful positioning, regular gentle range of movement exercises, joint injections, and medications [11,26]. The timely and proper use of assistive devices is also an important point of care, which includes the use of special mattresses and pillows, custom-fitted wheelchairs, and neutral-position splints for the hands and ankles to prevent joint contractures [64,65].
If pain is not controlled by positioning, splints, joint injections, or physical therapy, regular administration of analgesics should be considered using the World Health Organization analgesic ladder originally developed for the management of cancer-related pain, as suggested by the AAN Practice Parameters and EFNS guidelines [16,17]. However, the use of this strategy in patients with ALS is still debatable [67,68].
Conclusion
Pain is an important issue in ALS, with a pronounced impact on quality of life and suffering. The ultimate goal of pain treatment is to reduce its intensity, diminish suffering, and improve the quality of life of patients. To achieve this goal, thorough and timely assessments are vital. However, there is currently no assessment tool specific to this population. Therefore, a consensus should be reached regarding the timing, intervals, and contents of pain assessments in ALS. In addition, specific guidelines on the treatment of pain in ALS should be developed because successful pain treatment can improve the quality of life of patients and their caregivers, even in the absence of a disease cure. Further research on this topic is needed, considering the gaps between current care standards and patient requirements. In particular, careful consideration of the study design is required [26], considering that a classic double-blind, placebo-controlled study might not be feasible owing to the nature of the disease (rare, rapidly progressing, and incurable). Until more robust evidence is available, strategies for managing pain in ALS should be tailored to the needs of individual patients, based on good clinical practices and information from the management of nonmalignant chronic pain.
 XML Download
XML Download