

 PDF
PDF Citation
Citation Print
Print



Introduction
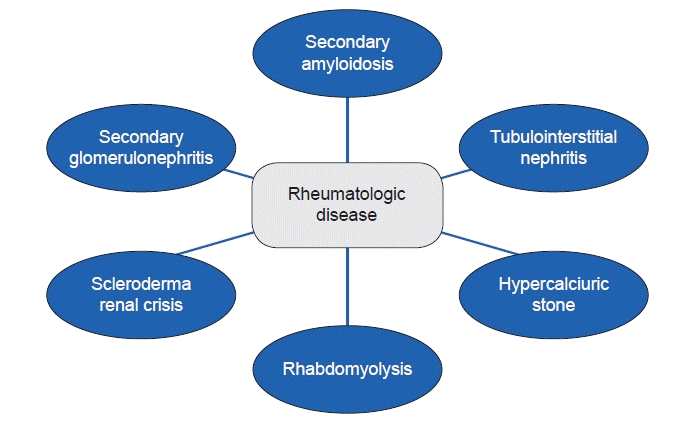
The kidneys are important target organs involved with systemic disease, which includes rheumatologic disease. The field of pediatric rheumatology originated in the first half of the 20th century and has a relatively short history compared to other medical fields. It started principally with interest in juvenile chronic inflammatory arthritis, the most common childhood rheumatologic disease [1]. Currently, its scope is expanding to address rare disease groups that have recently been elucidated, including autoinflammatory syndrome. Pediatric rheumatologic disease mainly involves various acute and chronic diseases targeting the musculoskeletal system, blood vessels, and other tissues, and is still a significant cause of chronic illness in children worldwide; although it remains among one of the smallest pediatric subspecialities [2]. Pediatric rheumatologic diseases are frequently associated with renal disease as a part of systemic autoimmune disease, and in some diseases such as systemic lupus erythematosus and antineutrophil cytoplasmic antibody-associated vasculitis, the kidney is the main target organ that can indicate the long-term prognosis. Renal manifestations in childhood rheumatologic disease vary from asymptomatic to end-stage kidney disease (Fig. 1). It is important to recognize that renal abnormalities can be a symptom of rheumatologic disease because they can provide important signals towards establishing a personalized treatment plan. In addition, kidney abnormalities may be a presenting symptom of rheumatologic disease; in this case, clinicians should attempt to identify the underlying disease. In this paper, we review kidney problems that can be accompanied by representative pediatric rheumatologic diseases, including juvenile idiopathic arthritis (JIA), Sjogren's syndrome (SS), systemic sclerosis/scleroderma, and juvenile dermatomyositis (JDM). Lupus nephritis and antineutrophil cytoplasmic antibody-associated renal disease tend to expand over a wide range of topics, and thus were excluded from this review.
Juvenile idiopathic arthritis
JIA is characterized by chronic noninfectious inflammation of the joints and encompasses a complex group of diseases. It is the most famous and frequent rheumatoid disease in children and is classified into several groups, according to clinical and laboratory characteristics. In the early history of JIA, gold nephropathy was an interesting kidney disease associated with the use of intramuscular gold salts, although there is currently no gold treatment. The pathological picture of gold nephropathy is drug-induced membranous glomerulonephritis that usually resolves over time if gold treatment is stopped [3,4].
Although it is difficult to determine the cause of renal abnormalities in JIA, according to one prospective study in adults, proteinuria and decreased renal function were mainly due to drug side effects, while hematuria was associated with the disease itself [5]. The renal diseases associated with JIA have rarely been reported, yet include renal amyloidosis, glomerulonephritis, and drug-induced tubulointerstitial nephritis (TIN). Amyloidosis is the most characteristic lesion associated with chronic systemic inflammation in JIA [6].
Renal amyloidosis
Amyloidosis is characterized by the deposition of amyloid fibrils in organs and there are a number of subtypes. Amyloid A (AA) amyloidosis is caused by the overproduction of the precursor of AA protein, which is produced in response to systemic inflammation, while amyloid light-chain amyloidosis is caused by the overproduction of monoclonal immunoglobulin light chains. Only AA amyloidosis (secondary amyloidosis) can occur in children with JIA [7,8]. In the past, amyloidosis was the main cause of death in JIA; however, recently, the prognosis has improved [8-10]. Renal amyloidosis occurs most commonly in systemic onset JIA (sJIA), followed by polyarticular JIA [8,9]. Renal amyloidosis insidiously progresses, causing massive proteinuria from an asymptomatic state, and consequently leading to end-stage renal disease. Hematuria is rarely accompanied [8,11]. Regular urinalysis is required in patients with sJIA or polyarticular JIA as asymptomatic proteinuria is the most common initial symptom [7]. It can be confirmed by renal biopsy which demonstrates amyloid fibrils, although the correlation between the degree of amyloid deposition and clinical symptoms are not clear. Treatments that control the inflammatory cascade caused by the underlying diseases are critical. Several disease-modifying antirheumatic drugs and biologics have been used to control these diseases. Since renal amyloidosis occurs in a situation where JIA is not well controlled by standard drugs such as methotrexate, sulfasalazine, and hydroxychloroquine, it is usually treated by adding disease-modifying antirheumatic drugs or biologics or switching biologics after the diagnosis of amyloidosis [12]. Interleukin-6 inhibitor, tocilizumab has become the standard treatment for sJIA, and it can play an important role in treating secondary amyloidosis by suppressing serum AA levels [13,14].
According to one large study conducted in 2008, of the 3,500 patients with JIA, 24 patients with biopsy-proven amyloidosis were detected. Ten patients died, but the cause of death was associated with JIA itself rather than with amyloidosis. Of the 14 survivors, three patients underwent kidney transplants, and 11 patients maintained normal renal function at last follow-up. Proteinuria improved completely in four patients who initially had proteinuria [8]. Renal disease can be improved by early intensive treatment. Therefore, it is essential to monitor regularly the occurrence of amyloidosis in JIA patients.
Glomerulonephritis and drug-induced TIN
Several studies on adult rheumatoid arthritis (RA) suggest that there is an association between RA and different types of glomerulonephritis, although studies on glomerulonephritis in JIA are extremely rare. Mesangial proliferative glomerulonephritis is the most commonly reported type of glomerulonephritis in adults with RA [15]. In JIA, membranous nephropathy, mesangial glomerulonephritis, focal segmental glomerulosclerosis, and crescentic glomerulonephritis have also reported [15-21]. Nephrotic syndrome in JIA is extremely rare and is usually caused by amyloidosis rather than glomerulonephritis [20,22]. The pathogenesis of renal involvement in JIA remains unclear. Immunologic abnormalities related to the occurrence of JIA, including hypergammaglobulinemia, abnormal B and T cell mitogen responsiveness, decreased T suppressor activity, and uncontrolled cytokine production, are presumed to lead to renal involvement [23,24]. In JIA, the treatment for glomerulonephritis is generally conservative with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers. However, severe cases with rapidly progressive glomerulonephritis or massive proteinuria require intensive treatments with immunosuppressants [16-22]. One should be aware that drugs used as treatments can also cause renal abnormalities. Drugs commonly used in JIA include nonsteroidal anti-inflammatory drugs, proton pump inhibitor, methotrexate, sulfasalazine, leflunomide, etc. Since these drugs can often cause renal abnormalities such as acute tubular necrosis and TIN. Clinicians should be careful to identify the cause of renal abnormalities in patients taking these drugs.
Sjogren's syndrome
Primary SS is a chronic autoimmune disease characterized by inflammation of exocrine glands including salivary and lacrimal glands [25]. In addition, it can demonstrate various exocrinopathy involving respiratory, urogenital tract and skin. Furthermore, extraglandular and systemic symptoms can also be accompanied [26]. SS is typically classified as primary or secondary. Primary SS has no association with other autoimmune diseases, while secondary SS has another underlying or combined autoimmune disease such as systemic lupus erythematosus, RA, and mixed connective tissue disease [27]. The suggested pathogenesis is that genetically susceptible individuals are exposed to environmental factors such as infection, leading to inflammatory processes in target tissues [28]. Definite diagnosis of SS is difficult, because cardinal symptoms in SS can be commonly seen and there is no diagnostic gold standard test. Clinicians generally use classification criteria for diagnosis, such as rheumatologic diseases. Autoantibodies to nuclear antigens Ro/SSA and La/SSB are the hallmarks of SS, but typically are not present in 30% to 50% of SS.
Renal involvement is uncommon in patients with SS; 5% to 33% of adult SS patients and approximately 10% of pediatric SS patients having renal involvement [29,30]. TIN is most frequently reported. TIN usually results from activated lymphocytic infiltration (primarily CD4+ T lymphocyte) to tubular epithelium and interstitium around the renal tubules, and this pathophysiology is similar to the process that occurs in exocrine glands [31]. TIN presents tubular dysfunction including renal tubular acidosis (mostly distal type but, proximal or mixed type is also possible), renal Fanconi syndrome, Bartter and Gitelman syndromes and nephrogenic diabetes insipidus [30,32-36]. Hypokalemia is a general symptom in SS-related renal disease, which is observed in about 40% of patients [32]. Actually, rheumatologic diseases are the second common cause of TIN accounting for 10% to 20% of all TIN cases [37]. Several rheumatologic diseases associated with TIN are described in Table 1.
Glomerular disease is less prevalent than tubular disease, and its pathophysiological process is mediated by the immune complex, while TIN is mediated by direct lymphocytic infiltration. Membranoproliferative glomerulonephritis due to cryoglobulinemia is the most commonly reported condition. In a study of 22 children with SS, 13 children showed renal involvement, only three of whom had glomerulonephritis, including mesangial proliferative glomerulonephritis, immunoglobulin A (IgA) deposits, membranous glomerulonephritis, and pauci-immune crescentic glomerulonephritis [29,38-40].
Immunosuppressants are not universally required in patients with SS; however, immunosuppressants such as steroid may be helpful in cases of severe systemic symptoms, including arthritis, fatigue, and recurrent parotitis [28]. Patients with symptomatic TIN also require treatment. Patients with TIN associated with SS generally respond well to steroid alone. In cases of steroid dependency, other immunosuppressants may be effective in reducing steroids. Steroid with rituximab and plasmapheresis are effective [25].
It is important to monitor for renal involvement in known SS patients, and it is also important to consider SS as the cause in patients with renal disease, primarily TIN, especially for nephrologists. However, it is challenging because symptoms such as sicca are insidious and commonly unclear [30]. In this case, high serum immunoglobulin G (IgG) level, positive rheumatoid factor, positive anti-Ro, anti-La, very high erythrocyte sedimentation rate, and cryoglobulins are clues suggesting SS [28,41]. In a previous adult study, long-term renal prognosis appeared to be good, however, the risk of chronic kidney disease is significantly elevated relative to the general population [42].
IgG4-related disease
IgG4-related disease (IgG4-RD) is a new disease entity that has emerged in the last decade and shares several common symptoms with SS, such as glandular enlargement, sicca, arthralgia, and high levels of IgG [43]. It is characterized by highly increased levels of IgG4 (>140 mg/dL) and striking IgG-producing plasma cell infiltration in the affected organ. The clinical manifestations varied widely. Diagnosis is usually made by histological examination demonstrating a classic fibrotic lesion with IgG4+ plasma cells. IgG4-RD is rare, occurring mainly in middle-aged men. TIN is the most common renal disease in IgG4-RD. Obstructive acute kidney injury (AKI) caused by retroperitoneal fibrosis can occur [44]. The renal IgG4-RD in children has been reported very limitedly. In most cases, it occurred concomitantly with other organ manifestations, except for one isolated renal IgG4 pseudotumor [45,46]. The use of steroid tends to be effective in most cases. IgG4-RD needs to be considered in cases with TIN accompanied by involvement of several organs, although it is rare.
Systemic sclerosis and localized scleroderma
Scleroderma disorders include both systemic sclerosis (SSc) and localized scleroderma (LS). LS is mainly restricted to the skin, whereas SSc affects the skin, vessels, and internal organs, including the gastrointestinal, pulmonary, and musculoskeletal organs [47]. It is an autoimmune disease characterized by vasculopathy and fibrosis. Renal involvement is uncommon in both SSc and LS, although mild renal dysfunction commonly occurs due to vasculopathy in SSc with a close frequency in children and adults with SSc. The most specific renal involvement is scleroderma renal crisis (SRC), which is more common in SSc. It is characterized by progressive AKI with severe hypertension, microangiopathic hemolytic anemia, and thrombocytopenia and renal involvement may continue asymptomatic until the late stages. In the past, it was a major cause of death, but recently, with appropriate treatment, the mortality rate has decreasing [22,48,49]. SRC has rarely been reported in children with SSc [47]. The pathogenesis of SRC still remains elusive, but the essential process is suspected to be injury of endothelial cells, leading to intimal thickening and proliferation of branched renal arteries.
The common histologic finding is an onion skin lesion of the renal interlobular artery. Additionally, episodic vasospasm in cortical arteries contributes to renal ischemia or hypoperfusion and activation of the renin-angiotensin system [48,50]. This mechanism is often called the renal Raynaud phenomenon, in connection with the fact that Raynaud phenomenon in fingers and toes is a typical and essential symptom in SSc. Early recognition of SRC is essential for its management. In adult guidelines, ACE inhibitors are recommended as the first-line treatment. If treatment is delayed, there is a possibility of irreversible kidney damage and death [51]. Special attention should be drawn to the development of SRC in patients taking steroid since studies have demonstrated an association between SRC and steroid in adult. In severe cases, treatment with eculizumab, a C5 blocker, may be needed, similar to refractory systemic thrombotic microangiopathy [48]. The effect of prophylactic ACE inhibitor is not evident. Although, compared to other rheumatologic diseases, secondary amyloidosis is very rare in SSc, it should be considered in cases of long-standing and progressive SSc with proteinuria [52].
Juvenile dermatomyositis
JDM is a rare systemic autoimmune disease mainly affects skin and muscles and accounts for 80% to 85% of all inflammatory myopathies in children. It is characterized by proximal muscle weakness and typical skin rashes, such as heliotrope rashes and Gottron papules. Other organs can be involved including the lungs, heart, gastrointestinal tract, and kidneys. Constitutional symptoms such as fever, fatigue, anorexia, and weight loss are common, and the onset is usually insidious. Histological findings are characterized by vascular and perivascular inflammation, and vasculopathy is considered the key to the pathogenesis of myositis and cutaneous symptoms. It is also associated with other manifestations including intestinal perforation, ulcerations, pulmonary disease, and cutaneous calcinosis [53]. The spectrum of renal disease in inflammatory myopathies in adults includes AKI, chronic kidney disease, glomerulonephritis, myoglobinuria, and hypertension, while data on children with JDM are lacking [54]. Membranous nephropathy was the most common chronic renal sequela in adults with inflammatory myopathy [55]. Few case reports in JDM demonstrated IgA nephropathy and nephrotic syndrome with AKI.
The children responded well to treatment with steroids and methotrexate for the primary disease, JDM, and usually do not require additional drugs for renal involvement except ACE inhibitors or angiotensin receptor blockers. However, in cases of IgA nephropathy with nephrotic-range proteinuria or progressive AKI, additional immunosuppressants such as cyclosporin, mycophenolate mofetil, azathioprine, and cyclophosphamide should be considered [56,57].
Rhabdomyolysis with AKI is extremely rare in JDM, but can occur especially in fulminant JDM with multiorgan involvement [58]. Macrophage activation syndrome may occur as a result of excessive systemic inflammation in JDM such as sJIA, and one case of thrombotic microangiopathy secondary to macrophage activation syndrome in JDM has been reported [59].
Conclusions
Rheumatologic diseases in children can affect various organs, including the musculoskeletal, cutaneous, pulmonary, heart, gastrointestinal, central nervous system, and kidneys. It is necessary to understand the type of renal disease associated with each rheumatologic disease to properly monitor and treat renal involvement in patients with rheumatologic diseases. In certain cases where patients are initially expressing symptoms of renal disease alone, we should try to find the underlying diseases, and then renal disease can be a diagnostic clue for underlying rheumatologic diseases.
 XML Download
XML Download