

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The endothelium refers to the monolayer of endothelial cells in the lumen of blood vessels that regulates the movement of blood components into surrounding tissues [1]. Disruption of endothelial function by excessive inflammatory and oxidative stress increases the adhesion and infiltration of immune cells and proliferation of vascular smooth muscle cells into blood vessel walls, which can result in plaque formation and cardiovascular disease (CVD) [2]. These processes are involved in various metabolic diseases such as obesity and type 2 diabetes mellitus (T2DM) [2].
G protein-coupled receptor 40 (GPR40), a receptor for medium and long chain fatty acids [3], has received attention as a therapeutic target for T2DM. GPR40 couples with a G protein subunit Gαq that can increase glucose-stimulated insulin secretion via regulation of intracellular calcium level in the pancreas [4]. Consequently, treatment with GPR40 agonist [5] or pancreatic GPR40 overexpression [6] increased plasma insulin level and improved glucose tolerance. GPR40 activation has been shown to be effective for preventing several metabolic diseases as well as T2DM. Li et al. [7] reported that GPR40 agonism reduced lipogenic gene expression and triglyceride accumulation via adenosine monophosphate-activated protein kinase phosphorylation in the liver of mice fed a high-cholesterol diet. Omega-3 fatty acids inhibited nucleotide oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation, which is involved with insulin resistance, pro-inflammatory cytokine expression, and caspase 1 cleavage, via GPR40 [8]. Additionally, ultraviolet-induced reactive oxygen species production, cytotoxicity, and inflammatory actions were attenuated after treatment with GPR40 agonist [9]. These reports suggest that GPR40 agonism is a novel therapeutic strategy that can be applied to overcome inflammatory stress. Therefore, we hypothesized that treatment with GPR40-specific agonists could attenuate endothelial inflammation.
METHODS
Cell culture and reagents
Human umbilical vein endothelial cells (HUVECs, Thermo Fisher Scientific, Waltham, MA, USA) were maintained in 0.2% gelatin-coated dishes with phenol red-free Medium 200 (Thermo Fisher Scientific) containing Low Serum Growth Supplement (Thermo Fisher Scientific) and was used at passages 3 to 6 for this experiment. THP-1 cells (Korean Cell Line Bank, Seoul, Korea) were incubated in RPMI 1640 (Thermo Fisher Scientific) containing 10% fetal bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin (Thermo Fisher Scientific). HUVECs were cultured to above 80% to 90% confluence before treatment with GPR40 agonists (HyunDaiPharm Ltd., Seoul, Korea) or other additives. The GPR40 agonists LY29-22470 and TAK875 and the GPR40 antagonist GW1100 (Cayman Chemical, Ann Arbor, MI, USA) were dissolved in dimethylsulfoxide (DMSO, Sigma Aldrich, St. Louis, MO, USA), and lipopolysaccharide (LPS, Sigma Aldrich) was diluted in phosphate-buffered saline. This article does not contain examinations performed on human participants. Then, ethical approval was not necessary.
Western blot
Proteins were separated in polyacrylamide gel and transferred to a nitrocellulose membrane (Amersham Bioscience, Waltham, MA, USA). The membranes were reacted sequentially with 5% non-fat dry milk, primary antibody, and secondary antibody in Tris-buffered saline containing 0.05% Tween 20 (TBST buffer). The reacted membranes were developed using ECL Western blotting solution (Bio-Rad Laboratories, Hercules, CA, USA).
Cell to cell adhesion assay
The adhesion abilities of HUVECs and THP-1 cells were determined using a green fluorescence dye, 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF/AM, Thermo Fisher Scientific). The detailed process was described in our previous report [10].
Immunofluorescence microscopy
HUVECs were fixed with 3.8% formaldehyde and permeabilized with pre-chilled MeOH for 10 minutes at −20°C. The cells were reacted sequentially with 5% non-fat dry milk, primary nuclear factor-kappa B (NF-κB) antibody, and secondary red fluorescence dye-labeled antibody in TBST buffer. The nuclear area was stained by Hoechst dye (blue). The stained cells were observed under the fluorescence microscope.
RESULTS
GPR40 agonism inhibited LPS-mediated NF-κB activation in HUVECs
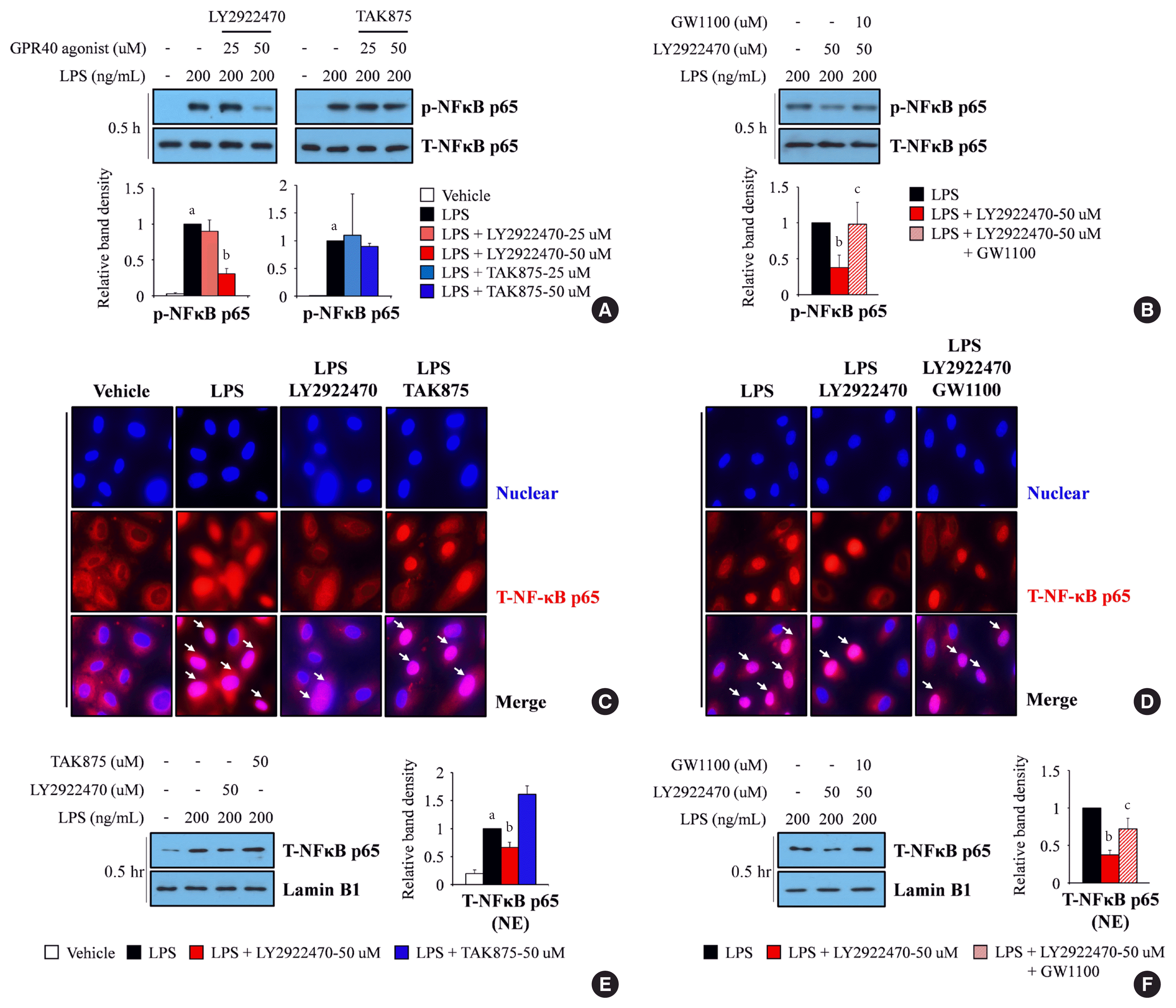
Western blot assay showed that LPS-mediated NF-κB phosphorylation was reduced dramatically after treatment with LY2922470 but not with TAK875 (Fig. 1A). The decrease in phosphorylated NF-κB level mediated by LY2922470 was restored after GW1100 treatment (Fig. 1B). Through immunofluorescence staining, we determined that NF-κB translocation from cytosol to nucleus was countered by LY2922470 under LPS treatment (Fig. 1C), and the translocation was reversed after GW1100 treatment (Fig. 1D). In addition, we rechecked NF-κB location by using nuclear fractionation assay kit, suggesting that LPS-induced nucleus-located NF-κB level was reduced after LY2922470 treatment (Fig. 1E), and the reduction was canceled by GW1100 (Fig. 1F). However, TAK875 did not inhibit LPS-mediated NF-κB movement (Fig. 1C and E).
GPR40 agonism attenuated LPS-induced expression of adhesion molecules in HUVECs
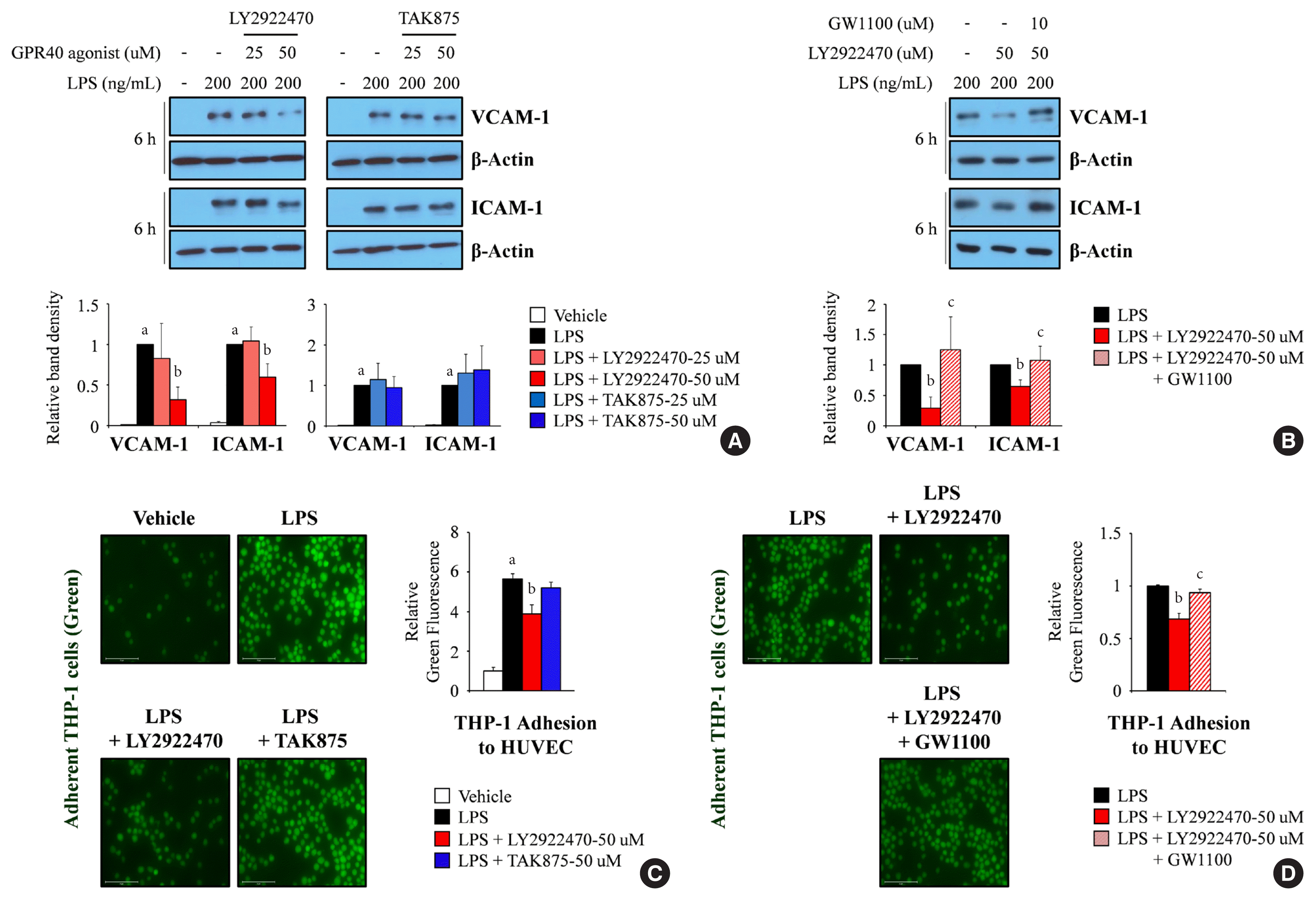
The results indicated that LY2922740 significantly inhibited LPS-mediated expression of adhesion molecules, specifically vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) (Fig. 2A), and these decreases were significantly reversed by GW1100 (Fig. 2B). In addition, LPS-mediated attachment of THP-1 cells to HUVECs was attenuated after treatment with LY2922470 (Fig. 2C), and the decreased attachment was restored after GW1100 treatment (Fig. 2D). However, TAK875 did not reduce the expression of adhesion molecules or attachment of THP-1 cells to HUVECs under LPS treatment (Fig. 2A and C).
DISCUSSION
GPR40-mediated signaling cascades and biological events are regulated by different ligands, because GPR40 has multiple ligand binding sites and interacts with various intracellular proteins, such as G protein Gαq, G protein Gαs, and β-arrestin 1/2 [11,12]. Hauge et al. [13] reported that the GPR40 agonists AM1638 and AM5262 stimulated G protein Gαq- and Gαs-induced signal pathways that resulted in dramatic elevation of incretin secretion. However, TAK875 stimulated only the G protein Gαq-mediated signal pathway and resulted in limited incretin secretion. They found that this difference was due to the diversity of the GPR40-ligand binding mode [13]. In the pancreas, TAK875 can enhance insulin secretion through a β-arrestin 2-dependant mechanism but not through palmitate and oleate, which are natural GPR40 agonists [12]. These ligand-specific responses are referred to as the ‘biased agonism’ of GPR40. In endothelial cells, GPR40-mediated effects were modulated differently by different ligands. TAK875 did not have any protective functions against LPS-induced inflammatory stress, whereas LY2922470 significantly inhibited LPS-mediated expression of adhesion molecules (Fig. 2A) and adhesion between THP-1 cells and HUVECs (Fig. 2C). In contrast, several studies have indicated that GPR40 is a metabolic stressor. Lu et al. [14] showed that palmitate accelerated LPS-mediated endothelial inflammation via GPR40. In the pancreatic β-cell line, GPR40 inhibition had a protective role against palmitate-mediated endoplasmic reticulum stress and cytotoxicity [15]. Therefore, we hypothesized that GPR40 can modulate the inflammatory response in a ligand-dependent manner.
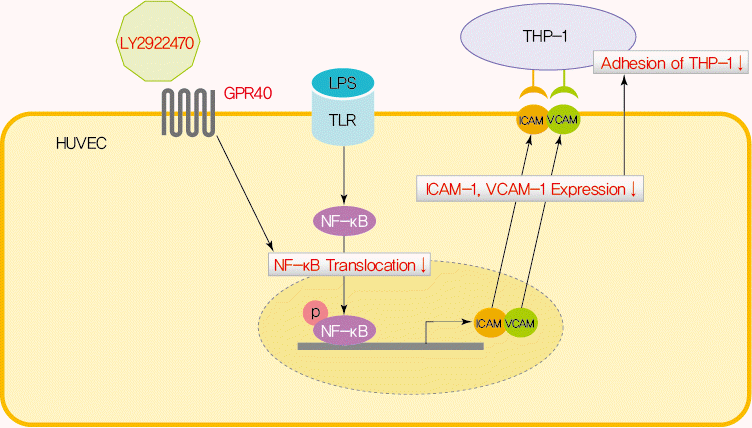
NF-κB is a major regulator of atherosclerotic processes [2]. Persistent and excessive NF-κB activation strongly enhances the expression of vascular adhesion protein and induces immune cell adherence and infiltration into the arterial intima layer. The results indicated that NF-κB inhibition reduced adhesion molecule expression, monocyte infiltration, and overall atherosclerotic plaque area [16,17]. In this experiment, we showed that treatment with a GPR40-specific agonist, LY-2922470, significantly decreased LPS-mediated NF-κB phosphorylation (Fig. 1A) and translocation to the nucleus (Fig. 1C and E), which resulted in attenuated adhesion of THP-1 cells to HUVECs (Fig. 2C). These protective effects were attenuated by treatment with GW1100, a GPR40 antagonist (Figs. 1F and 2D). Our results indicate that GPR40-mediated inhibition of NF-κB activation in endothelial cells can translate to inhibition of atherosclerotic processes.
We describe here for the first time that endothelial GPR40-specific agonism can modulate inflammatory responses in a ligand-dependent manner. LY2922470 inhibited LPS-mediated NF-κB activation and endothelial cell monocyte adhesion, but TAK875 did not. Although clinical development of TAK875 was terminated during phase III trials because of liver toxicity [18], we propose that biased agonism of GPR40 represents a good therapeutic target for impacting metabolic diseases such as T2DM and CVD.
 XML Download
XML Download